12. Tumeurs stromales gastro-intestinales (GIST)
(Dernière mise à jour le : )Principaux changements des mises à jour
Principaux changements de la mise à jour du 11/06/2024
- Modification complète de l’architecture du chapitre avec 4 sous-chapitres pour un accès plus immédiat aux informations essentielles : 12.2. bilan initial ; 12.3. GIST localisées ; 12.4. GIST avancées ; 12.5. indications thérapeutiques
- 12.3.3.1 et 12.5.1. GIST gastriques < 3 cm : prise en charge endoscopique
- 12.2.3. et 12.4.3.3. données de biologie moléculaire
- 12.3.6.2., 12.5.1. et annexe 3. nouvelle durée standard de 6 ans imatinib traitement adjuvant : essai IMADGIST 3 ans vs 6 ans
- 12.4.3.3. et 12.4.3.6. GIST avancées : résultats ADNtc essai INTRIGUE avec riprétinib
- 12.4.3.7. GIST avancées PDGFRA D842V : remboursement de l’avapritinib
- 12.5.6 à 9. actualisation des essais cliniques disponibles :
• PRODIGE 92 - TARGET MONITO DIG
• GIST-TEN
Principaux changements de la mise à jour du 23/10/2024
- 12.2.4. et 12.3.3.3. GIST en lien avec NF1 : intégration des données de l’étude RECKgist avec score de risque de récidive
- 12.4.3.6. et 12.5.9. remboursement du riprétinib
- 12.4.3.10. et 12.5.9. autres TKI au-delà de la 3ème ligne : option du lenvatinib (essai LENVAGIST).
Groupe de travail et relecteurs
Groupe de travail :
Marc PRACHT (Coordonnateur, Rennes),
Jean-Yves BLAY (Lyon), Sylvie BONVALOT (Paris), Olivier BOUCHE (Reims), Florence DUFFAUD (Marseille), Jean-François EMILE (Boulogne-Billancourt), Vincent HAUTEFEUILLE (Amiens), Charles HONORE (Villejuif), Widad LAHLOU (Paris), Eric LARTIGAU (Lille), Valérie LAURENT-CROISE (Nancy), Axel LE CESNE (Villejuif), Bruno LANDI (Paris)
Relecteurs :
T APARICIO (Paris), L BENHAIM (Villejuif), M BRAHMI (Lyon), M BRASSEUR (Reims), S. CATTAN (Lille), JM COINDRE (Bordeaux), Michel DUCREUX (Villejuif), N FIRMIN (Montpellier), S GAUJOUX (Paris), F HUGUET (Paris), L. MOSSER (Rodez), N PENEL (Lille), S SALAS (Marseille), N STOCK (Rennes), T LECOMTE (Tours)
Comment citer ce chapitre ?
Gastrointestinal stromal tumours (GISTs): French Intergroup Clinical Practice Guidelines for diagnosis, treatments and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO)
Landi B, Blay JY, Bonvalot S, Brasseur M, Coindre JM, Emile JF, Hautefeuille V, Honore C, Lartigau E, Mantion G, Pracht M, Le Cesne A, Ducreux M, Bouche O; Thésaurus National de Cancérologie Digestive (TNCD). Dig Liver Dis. 2019 Sep;51(9):1223-1231.doi: 10.1016/j.dld.2019.07.006.
Et mise à jour 2024 :
Pracht M, Blay JY, Bonvalot S, Duffaud F, Emile JF, Hautefeuille V, Honore C, Lahlou W, Lartigau E, Laurent-Croise V, Le Cesne A, Landi B, Ducreux M, Bouché O «Tumeurs Stromales Gastro-Intestinales (GIST)». Thésaurus National de Cancérologie Digestive, octobre 2024, en ligne http://www.tncd.org
Recommandations sous égide du
Groupe Sarcome Français (GSF-GETO/NetSarc+)

Les associations nationales ou internationales de patients jouent un rôle essentiel dans la diffusion de l’information et dans l’aide à la prise en charge de ces tumeurs rares.
L’association Info Sarcomes est l’association française des patients porteurs de GIST : https://www.infosarcomes.org/
12.1. Introduction
12.1.1. Méthodologie
Ce travail est le fruit d’une synthèse issue des données de la littérature faite par un groupe de travail représentant plusieurs sociétés savantes : la Fédération Francophone de Cancérologie Digestive (FFCD), de la Fédération Nationale des Centres de Lutte Contre le Cancer (UNICANCER), le Groupe Coopérateur multidisciplinaire en Oncologie (GERCOR), la Société Française de Chirurgie Digestive (SFCD), de la Société Française d’Endoscopie Digestive (SFED), l’Association Française de Chirurgie (AFC) de la Société Française de Radiothérapie Oncologique (SFRO), la Société Française de Pathologie (SFP), la Société Nationale Française de Gastroentérologie (SNFGE), de la Société Française de Pathologie (SFP), et de la Société Française de Radiologie (SFR) ainsi que le Groupe Sarcome Français (GSF-GETO) et le réseau NetSarc+.
Il s’appuie sur les dernières recommandations publiées de l’ESMO (Casali 2022), du National Comprehensive Cancer Network (www.nccn.org) (Von Mehren, 2014 ; Von Mehren, 2022), ainsi que sur une actualisation des données par une recherche bibliographique.
Celle-ci a reposé sur l'extraction, à partir de la base de données PubMed interrogée en janvier 2024, des études cliniques et essais randomisés, méta-analyses et recommandations de pratique clinique avec les mots-clés « gastrointestinal stromal tumor », «GIST» sans limitation de date.Les présentes recommandations ont été gradées selon le niveau des preuves disponibles dans la littérature, correspondant à la présentation retenue pour le TNCD selon 4 niveaux (A, B, C, accord ou avis d’experts) résumés dans le tableau 1.
Les formes pédiatriques de GIST ne seront pas traitées ici.
Tableau 1
Système de gradation des recommandations utilisé dans ce chapitre
| GRADE | NIVEAU DE PREUVE CORRESPONDANT |
|---|---|
| A | Recommandation forte basée par exemple sur un/des essai(s) comparatif(s) randomisé(s) de forte puissance, une/des méta-analyse d’essai(s) comparatif(s) randomisé(s), ou une analyse de décision fondée sur des études bien menées. |
| B | Recommandation basée sur une présomption scientifique à partir d’essais comparatifs randomisés de faible puissance, d’études comparatives non randomisées bien menées ou d’études de cohortes. |
| C | Recommandation basée sur un faible niveau de preuve à partir d’études cas-témoins, d’études comparatives comportant des biais importants, d’études rétrospectives, de séries de cas, d’études épidémiologiques descriptives (transversale, longitudinale). |
| Accord ou Avis d’experts | Recommandation basée sur un accord d’experts ou un avis d’experts en l’absence de données suffisantes de la littérature |
12.1.2. Présentation et épidémiologie
Les GIST sont des tumeurs mésenchymateuses malignes se développant dans la paroi du tube digestif, le plus souvent dans l’estomac et le grêle, plus rarement dans le rectum, le côlon, l’œsophage (Miettinen, 2006 ; Soreide 2016). Sur le plan cellulaire, les GIST ont le phénotype des cellules interstitielles de Cajal, cellule ‘’pacemaker’’ à l’origine du péristaltisme digestif et située au sein de la couche musculeuse du tube digestif. (Corless, 2011).
Les GIST appartiennent à l’entité nosologique des sarcomes et en représentent environ 18% soit la forme la plus fréquente de sarcome (Ducimetiere, 2011). Leur incidence est estimée à environ 15 cas/million d'habitants/an soit près de 1000 nouveaux cas par an en France. L’âge médian au diagnostic est d'environ 60 ans et le sex-ratio est d’environ 1/1.
Les GIST sont diagnostiquées au stade localisé dans 80 à 90% des cas. Ce sont des tumeurs non lymphophiles (à l’exception des GIST syndromique en lien avec une perte d’expression de Succinate Deshydrogénase SDH, plus fréquentes dans les populations pédiatriques) mais ayant un tropisme de dissémination secondaire hépatique et péritonéal. (Corless, 2011).
Les GIST sont dans la très grande majorité des cas sporadiques ; les formes syndromiques sont à évoquer en cas d’âge au diagnostic inférieur à 40 ans et/ou en cas d’anamnèse familiale évocatrice. Ces dernières imposent une consultation d’oncogénétique (cf. 12.1.4.) (Kwak, 2023; Ricci, 2016).
Sur le plan anatomopathologique, les GIST montrent une différenciation fusiforme (70%), épithélioïde (10%) ou mixte (20%) sans impact pronostique ou thérapeutique majeur. En immunohistochimie, les GIST sont typiquement (98% des cas) de phénotype CD117/KIT+ ou DOG1+. Elles présentent très fréquemment (85 % des cas) des mutations activatrices des gènes codant pour les récepteurs à tyrosine-kinase KIT (75 à 80% des cas) ou PDGFRA (env. 10% des cas) (Corless, 2011). Les GIST constituent un ensemble hétérogène sur le plan de la biologie moléculaire, du comportement clinique et de la réponse au traitement.
12.2. Bilan diagnostique et pré-thérapeutique
12.2.1. Circonstances diagnostiques
Elles sont parfois fortuites (20% des cas) lors d’un examen scanographique abdominal, endoscopique digestif ou en peropératoire. Le plus souvent c’est un syndrome tumoral (douleur, masse palpable, déformation abdominale) ou une complication tumorale (perforation, hémorragie, occlusion) qui révèle la pathologie.
12.2.2. Biopsies
Les biopsies endoscopiques superficielles simples à la pince sont généralement négatives, car la tumeur se développe dans la musculeuse du tube digestif (Casali, 2022 ; Von Mehren, 2014).
L’indication d’une ponction-biopsie (par voie écho-endoscopique, percutanée ou encore chirurgicale) doit être discutée au cas par cas. Lorsqu’il est fait par voie percutanée ou cœlioscopique, un tel geste comporte un risque hémorragique et potentiellement de dissémination péritonéale.
Si la biopsie est recommandée en préopératoire de manière générale dans les sarcomes, elle n’est pas systématique en cas de GIST radiologiquement évidente mais indispensable en cas de doute diagnostique, de résécabilité incertaine, de chirurgie lourde ou mutilante et/ou de nécessité d’un traitement médical initial (requérant une analyse mutationnelle pour définir la sensibilité aux ITK). Cette biopsie peut alors être réalisée par écho-endoscopie pour les GIST gastroduodénales et rectales. Si une biopsie percutanée est nécessaire avant traitement, une aiguille coaxiale protégeant le trajet de ponction est recommandée avec dans la mesure du possible une préservation du péritoine.
REFERENCES
Si la tumeur paraît résécable, que le patient est opérable et qu’une chirurgie d’emblée est proposée (pas de modification du geste opératoire par un traitement par TKI) en réunion de concertation pluridisciplinaire (RCP), une biopsie préopératoire n’est pas obligatoire (accord d’experts).
La biopsie est recommandée si :
• Doute diagnostique avec une autre tumeur nécessitant une chimiothérapie première ou un geste chirurgical différent ou une stratégie thérapeutique alternative (par exemple : lymphome, autres sarcomes, tumeur desmoïde, tumeur neuro-endocrine...) (accord d’experts)
• Tumeur localement avancée/non-résécable d’emblée ou nécessitant une chirurgie mutilante ou faisant discuter un traitement néoadjuvant/ préopératoire par imatinib (accord d’experts).
• Tumeur métastatique (accord d’experts).
La biopsie peut porter sur la tumeur primitive ou sur une métastase : biopsie hépatique sous contrôle radiologique (échographie ou scanographie) ou écho-endoscopique. La ponction sous écho-endoscopie peut être privilégiée par rapport à la voie percutanée dans les GIST gastroduodénales ou rectales non métastatiques, même si dans l’essai randomisé du SSG XVIII/AIO, le risque de rechute des GIST de haut risque n’était pas augmenté par la biopsie préopératoire percutanée (Eriksson, 2016). La biopsie doit être réalisée en centre spécialisé quand le risque de complication est important (GIST kystiques ou hémorragiques).
La biopsie doit être de taille suffisante pour le diagnostic histologique et pour la recherche de mutation de KIT ou PDGFRA, lorsqu’elle est indiquée. Elle doit être fixée dans du formol à 4 %. Le génotypage peut se faire sur les fragments fixés au formol, la rentabilité est toutefois supérieure sur un fragment congelé.
12.2.3. Analyses histologiques et de biologie moléculaire
12.2.3.1. Histologie et immunohistochimie
Le diagnostic de GIST est effectué sur un examen histologique standard (10, 74).Casali, 2022 ; Von Mehren, 2014).
L’immunohistochimie (IHC) est nécessaire au diagnostic (accord d'experts). Les marqueurs indispensables sont KIT (CD117) et DOG-1 (pour Discovered On GIST), retrouvé chacun dans 95 % des GIST. En cas de négativité, d’autres marqueurs sont recommandés pour étayer le diagnostic (CD34, desmine, protéine S100) (accord d'experts). D’autres tumeurs, conjonctives, mélanocytaires ou endocrines, peuvent simuler une GIST. Les tumeurs les plus souvent confondues avec une GIST sont les fibromatoses / tumeurs desmoides et les métastases de mélanome. (Fletcher, 2002).
La double lecture des lames d’anatomopathologie en centre expert est recommandée par l’INCa pour tous les sarcomes dont les GIST (RRePS, réseau de référence en pathologie des sarcomes : https://rreps.sarcomabcb.org. Elle permet de limiter les erreurs diagnostiques ou celles en lien avec l’évaluation du risque de récidive et d’améliorer la prise en charge thérapeutique. Elle a en outre l’avantage de favoriser le génotypage des GIST.
12.2.3.2. Génomique
La recherche de mutations des gènes KIT et PDGFRA par une technique de biologie moléculaire est de pratique courante dans la prise en charge des GIST du fait de son intérêt théranostique. Le génotypage des GIST est recommandé à l’exception des GIST à très bas risque de récidive sans indication d’imatinib (cf. 12.2.5.). Le génotypage permet aussi de confirmer le diagnostic dans les cas difficiles de GIST KIT et/ou DOG-1 négatifs en IHC digestif (Casali, 2022 ; Von Mehren, 2014).
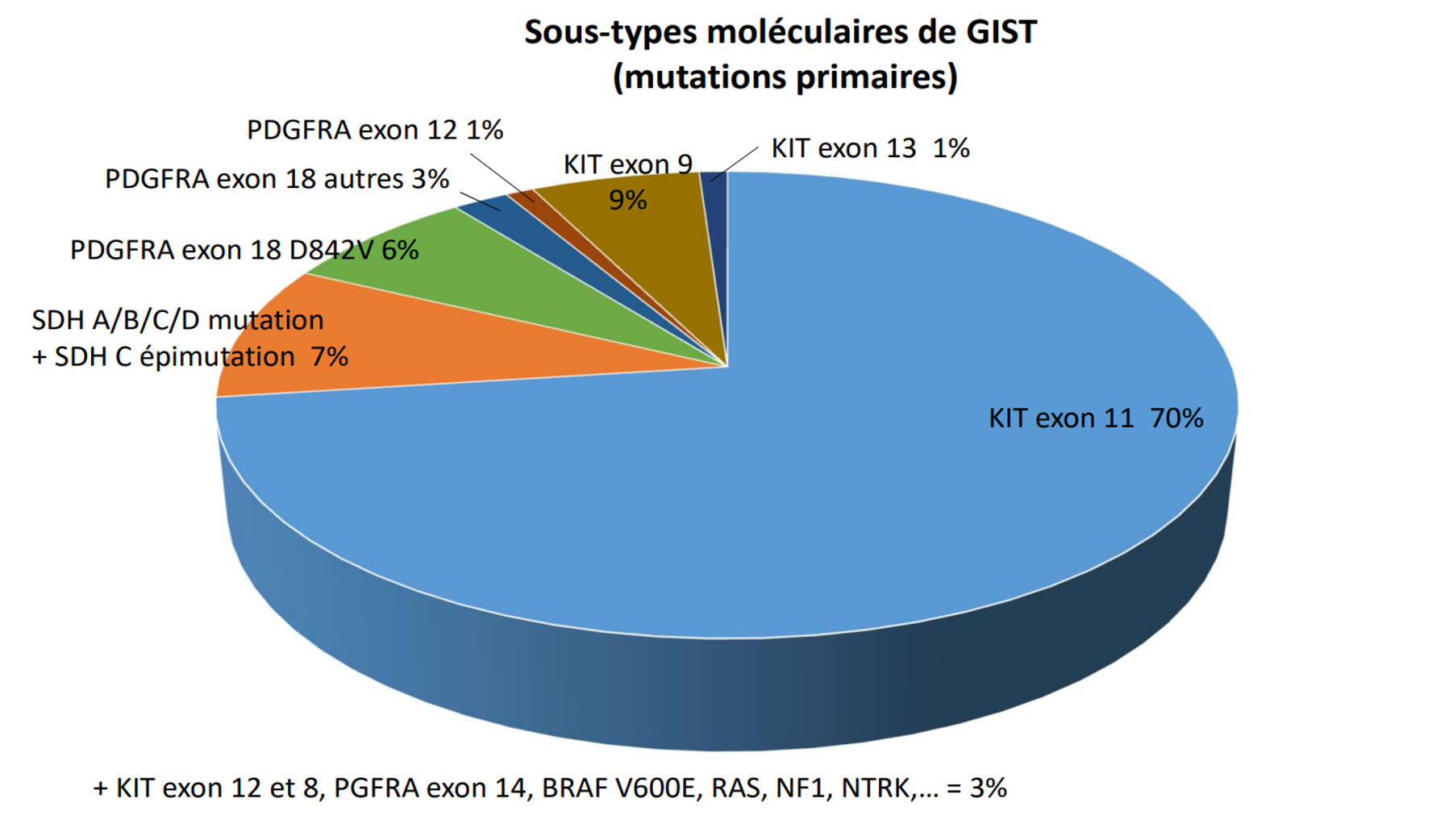
Le type de mutation a une influence sur le pronostic et l’efficacité du traitement en situation néoadjuvante, adjuvante et métastatique (Casali, 2022) (cf. 12.2.5.2.). Les mutations de KIT (environ 75-80% des cas) sont variables dans leur localisation et leur nature (délétions, duplications, substitutions …). Au diagnostic, la mutation primitive de KIT siège le plus souvent sur l’exon 11 (environ 70 % des cas). Les autres mutations primitives de KIT siègent sur l’exon 9 (10 à 15 % environ des GIST métastatiques) et plus rarement (<5%) sur d’autres exons (exon 13 pour 1 à 3 % et 17 pour 1 %). Les mutations de PDGFRA (10 % environ des GIST) siègent le plus souvent sur l’exon 18 (62%). La substitution p.D842V de l’exon 18 de PDGFRA rend la GIST primitivement résistante à l’imatinib (cf. 13.2.5.2.) ; elle représente environ 50 % des mutations de l’exon 18 et est particulièrement retrouvée dans les GIST gastriques (Trent, 2023).
Dans 10-15% des cas environ, on ne retrouve pas de mutation de KIT ou de PDGFRA. Ces GIST qui étaient regroupées sous le terme « wild type » (WT) correspondent en fait à un groupe hétérogène, où d’autres anomalies moléculaires sont souvent retrouvées : mutations des gènes NF1, BRAF, KRAS, NRAS, ATRX, PTEN, mutations ou pertes d’expression de sous-unités de SDH, fusions de gène NTRK (Nannini, 2017 ; Nishida 2024).
Ces GIST sont différentes sur le plan clinique, moléculaire, pronostique, et ne répondent pas forcément aux traitements des formes ‘’classiques’’ de GIST mutées sur KIT ou PDGFRA. Une IHC pour recherche d’une perte d’expression de SDH-B (sous-unité B de la succinate déshydrogénase) est recommandée en 1ère intention en cas de GIST sans mutation de KIT ou PDGFRA (Gasparotto, 2017). En effet, en cas de négativité, la recherche de métastases ganglionnaires, d’une forme syndromique et un conseil génétique peuvent être discutés (accord d’experts) (Casali, 2022).
Une recherche de fusion du gène NTRK (sélection par test IHC et confirmation par séquençage) (Marchio, 2019) et de mutation de NF1, BRAF, KRAS, NRAS est recommandée dans un 2ème temps pour les GIST avancées KIT et PGFRA wild type et non SDHB-déficiente en IHC (Fig. 1) (avis d’experts).
Figure 1
Sous-types moléculaires de GIST (mutations primaires) d’après (Trent, 2023)
La collection de tissu congelé doit être encouragée, de nouvelles évaluations moléculaires pouvant être utiles au cours de l’évolution de la maladie et dans le cadre de la recherche clinique (Casali, 2022). La création de tumorothèques est souhaitable.
A noter qu’une place grandissante est en train d’être faite à la détermination des néo-mutations de KIT par la re-biopsie ou par analyse de l’ADN tumoral circulant car elle pourrait permettre de choisir le traitement de seconde ligne et plus des GIST avancées.
12.2.4. GIST syndromiques
En l'absence de mutation de KIT ou PDGFRA, une IHC SDH-B est recommandée en 1ère intention. En effet, une perte d'expression de SDH-B fera rechercher une mutation de l'une des sous-unités de SDH ou d’une altération épigénétique responsable d’un défaut d’expression. Ces mutations ou altérations peuvent être constitutionnelles et donc orienter vers une consultation d'oncogénétique (accord d’experts). Par ailleurs les GIST avec perte d'expression de SDH-B peuvent donner des métastases dans les ganglions lymphatiques de drainage (Gasparotto, 2017 ; Ricci, 2016).
GIST SDH-dépendantes (très rares) :
• GIST sans mutation somatique de KIT et PDGFRA, cellules épithélioïdes, sujet jeune, antécédent familial de GIST. Intérêt de l’immunohistochimie SDH-B.
• Syndrome (ou dyade) de Carney-Stratakis par mutation constitutionnelle des gènes de la sous-unité A, B, C ou D de SDH : sans prédominance de genre, GIST gastriques multiples, le plus souvent épithélioïdes, parfois lymphophiles, paragangliomes extra-surrénaliens (pas de chondrome pulmonaire).
• Triade de Carney: pas de mutation constitutionnelle mais déficit (méthylation ou perte chromosomique) de SDH (méthylation promoteur gène SDH-C le plus souvent), femme jeune (âge médian diagnostic environ 20 ans), 3 entités synchrones ou métachrones : GIST gastriques multiples parfois lymphophiles, chondrome pulmonaire et paragangliome extra-surrénalien ;Neurofibromatose de type 1 : mutation constitutionnelle du gène NF1, les mutations somatiques des gènes KIT et PDGFRA étant très rares (5 % dans la cohorte RECKGIST), GIST très souvent multiples (59 % des cas), prédominant dans l’intestin grêle et touchant l’estomac dans 5 % des cas, de petites tailles et de bon pronostic ; tableau typique de NF1 associé (Gasparotto, 2017 ; Yegin, 2016)
Mutation constitutionnelle/germinale de KIT ou PDGFRA (exceptionnelle) : sujet jeune, antécédent familial de GIST, trouble de la motilité intestinale (dysphagie, constipation), tâches pigmentées cutanées, hyperplasie des cellules de Cajal (Bachet, 2013).
On recommande en cas de suspicion de mutation génétique constitutionnelle une consultation d’oncogénétique après information et accord du patient (accord d'experts). Les indications de la consultation d’oncogénétique et l’algorithme d’analyse génétique constitutionnelle en fonction de l’immunohistochimie SDH-B sont résumés dans l’annexe 1.
12.2.5. Bilan d'extension
Sur le plan morphologique, les GIST montrent sur les imageries en coupe un aspect homogène quand elles sont de petite taille et hétérogène (nécrose, hémorragie) pour les plus grosses. Elles ont un développement pariétal ou exoluminal, à contour régulier, à rehaussement périphérique et la plupart du temps sans extension ganglionnaire. De nombreux diagnostics différentiels radiologiques existent (lymphome, léiomyosarcome, desmoïde, adénocarcinome).
Sur le plan endoscopique, les GIST se présentent comme des lésions sous-muqueuses sans anomalie épithéliale, parfois ulcérées superficiellement ou responsable d’hémorragie digestive. En écho-endoscopie, elles sont typiquement développées dans la 4ème couche hypoéchogène (musculeuse), avec une échostructure hypoéchogène hétérogène. En Doppler, on note une vascularisation riche. Les biopsies avec aiguille coupante « histologiques » sont à préférer (avis d’experts) (Casali, 2022 ; Von Mehren 2014).
REFERENCE
• Scanner (TDM) thoraco-abdomino-pelvien trois temps (accord d'experts).
• Écho-endoscopie (avant une discussion chirurgicale ou de traitement néoadjuvant dans les GIST localisées du tractus digestif haut ou du rectum) (accord d'experts) .
OPTIONS
• IRM rectale pour les GIST du rectum afin d’évaluer l’envahissement des sphincters et des structures adjacentes (accord d'experts).
• IRM hépatique en cas de doute diagnostique sur des métastases (accord d'experts).
• TEP-Scan au 18FDG cas de doute sur une lésion métastatique en TDM ou IRM et en situation néoadjuvante (accord d'experts).
12.2.6. Bilan d'opérabilité
Orienté par les antécédents, l'examen clinique et la consultation d'anesthésie.
12.3. Prise en charge des GIST localisées
Le réseau de référence clinique NETSARC ( https://expertisesarcome.org ) labellisé par l'INCa comporte 25 centres spécialisés répartis sur tout le territoire national qui assurent la prise en charge de sarcomes des tissus mous et des viscères, des GIST et des tumeurs desmoïdes. Il est recommandé de présenter les dossiers de GIST à une RCP spécialisée d’un des centres.
12.3.1. Cas particuliers des GIST de taille < 2 cm
12.3.1.1. Estomac
Des séries suggèrent une prévalence élevée de GIST gastriques de petite taille (< 1 cm de grand axe) chez l’adulte de plus de 50 ans, dont l’évolutivité est incertaine voire qui pourraient régresser (Casali, 2022). La mutation de KIT est un phénomène précoce, mais d’autres anomalies seraient responsables de l’évolutivité de ces petites GIST, aussi appelées micro-GIST. Le risque d’évolution métastatique des GIST de l’estomac est très faible ou nul quand elles mesurent moins de 2 cm (cf. tableau du chapitre 12.3.5.1) possiblement lien avec une présence plus élevée de mutation de PDGFRA leur conférant une agressivité moindre (Yegin, 2016).
De fait, le choix entre surveillance ou résection est licite pour les GIST de l’estomac asymptomatique de moins de 2 cm, en tenant compte du terrain, de la localisation de la lésion dans l’estomac et de sa résécabilité attendue (simple ou complexe, la décision chirurgicale étant plus simple pour les GIST nécessitant une gastrectomie atypique que pour celles nécessitant une gastrectomie partielle ou totale (cf. Annexe 2). Dans tous les cas, une information et une discussion avec le patient sont nécessaires.
L’exérèse endoscopique de ces petites GIST gastriques par dissection sous muqueuse est une option (avis d’experts). Elle est alors à pratiquer dans des équipes expertes (cf. 12.3.3.1).
Aucun schéma de surveillance n’est validé en l’absence d’exérèse pour ces micro-GIST mais une surveillance par endoscopie ou mieux par écho-endoscopie à 6 mois, 18 mois puis tous les 2 ans, à adapter en fonction du contexte, semble un bon compromis entre sécurité et compliance (avis d’experts) (Landi, 2010). Après exérèse la surveillance sera adaptée au risque de récidive (cf. 12.2.5. et Annexe 3). Une surveillance par scanner avec préparation gastrique est proposée par certaines équipes.
12.3.1.2. Intestin grêle et rectum
Pour les GIST du rectum ou du grêle de moins de 2 cm de diamètre, la résection est la règle malgré une taille limitée du fait du risque évolutif (notamment si l’index mitotique est élevé : cf. tableau du chapitre 12.2.5) (Casali, 2022). Une histologie peut être nécessaire en préopératoire quand il existe un doute sur la nature exacte de la lésion notamment pour différencier un léiomyome d’une GIST (Jianchang, 2022) (avis d’experts).
Enfin, pour les GIST du rectum, une discussion de résection limitée par TEM ou DSM/FTRD pourrait également se justifier afin de privilégier la qualité de vie et limiter les séquelles fonctionnelles due à une chirurgie carcinologique du rectum.
12.3.1.3. Œsophage
Ces formes sont très rares et à discuter en RCP NETSARC+ pour les aspects thérapeutiques vu la morbidité du geste comme pour le schéma de surveillance. Une histologie peut là aussi être nécessaire en préopératoire quand il existe un doute sur la nature exacte de la lésion du 1/3 inférieur de l’œsophage ou de la jonction œsogastrique (léiomyome ?).
12.3.2. Principes de la chirurgie des GIST de plus de 2 cm
La résection chirurgicale complète en monobloc (sans effraction tumorale) de la tumeur sans curage ganglionnaire systématique est le traitement curatif de référence (Casali, 2022 ; Von Mehren 2014).
La cœlioscopie est licite si elle n’entraine pas de sur-risque d’effraction tumorale qui grèverait le pronostic du patient (perte de chance).
Il n’existe pas de consensus sur la marge optimale de résection, qui peut probablement être limitée, l’important étant une résection microscopiquement complète (R0) (accord d’experts).
Une chirurgie préservant les organes, c’est-à-dire sans interruption de la continuité digestive, peut être discutée si elle est réalisable avec une marge microscopiquement saine (notamment dans l’estomac).
Pour les GIST du bas rectum, de nombreuses séries rétrospectives montrent que si une exérèse locale par voie basse est possible sans effraction tumorale, la survie globale est la même que pour une résection rectale. Les énucléations ne sont pas recommandées, car classiquement grevées d’un risque de récidive plus élevé que les résections segmentaires, tout du moins pour les GIST gastriques et rectales (avis d’experts) (Jianchang, 2022).
Il est essentiel d’éviter une rupture tumorale préopératoire qui est un facteur prédictif quasi-absolu de récidive péritonéale malgré une chirurgie macroscopiquement complète, au point que certaines équipes recommandent un traitement adjuvant dans cette situation de rupture. En effet, le pronostic d’une rupture tumorale est le même que celui d’un patient métastatique. Les GIST volumineuses ou après traitement par ITK sont très fragiles, comprenant souvent des remaniements kystiques, hémorragiques et/ou nécrotiques. Elles doivent donc être manipulées avec la plus grande précaution (Casali, 2022 ; Von Mehren, 2014).
Il n’y a pas de définition consensuelle de la rupture tumorale dans les GIST. Un groupe de chirurgien a proposé sans qu’elles soient validées six situations à considérer comme une rupture tumorale: une effraction tumorale avec/sans essaimage (iatrogène ou spontanée), une ascite tachée de sang, une perforation gastro-intestinale au niveau de la tumeur (avec essaimage du contenu intra-luminal), une infiltration microscopique d'un organe de contact (T4b), une exérèse fragmentaire ou intra-lésionnelle, une biopsie chirurgicale par laparotomie ou laparoscopie (avis d’experts) (Nishida, 2019). Attention néanmoins à ne pas conclure à une GIST rompue en cas de grosse ulcération endoluminale, la rupture tumorale impactant la dissémination péritonéale uniquement lorsque la rupture est sur le versant péritonéal/exoluminal.
Le curage ganglionnaire n’est pas recommandé (hors infiltration ganglionnaire constatée en peropératoire), car les métastases ganglionnaires sont exceptionnelles et le risque de récidive ganglionnaire limité, sauf dans les formes pédiatriques et/ou liées à un déficit en SDH (accord d’experts). En cas de doute, un picking ganglionnaire est recommandé.
Après résection microscopiquement incomplète (R1), la conduite à tenir reste l'objet de discussions au cas par cas, car d’une part il n’a pas été démontré qu’une reprise améliorait le pronostic, et d’autre part, cette reprise n’est pas toujours un geste techniquement simple (Gronchi, 2020). Un traitement adjuvant sera à discuter selon les facteurs pronostiques associés (confer 12.3.6.2) comme pour les formes en résection complète (R0).
12.3.3. Chirurgie et traitements interventionnels des tumeurs localisées
12.3.3.1. Résection endoscopique
- Les équipes asiatiques pratiquent de plus en plus des résections endoscopiques pour les petites GIST gastriques (< 2 ou 3 cm), éventuellement par une approche combinée avec la cœlioscopie, mais cela reste à valider (Yegin, 2016).
- Dans ce cas, une résection par dissection sous-muqueuse ou FTRD (« full-thickness resection device ») est recommandée, car la GIST se développant dans la musculeuse et il est nécessaire de bien creuser dans cette dernière afin d’avoir une résection monobloc microscopiquement complète (R0). (avis d’experts).
12.3.3.2. Chirurgie des formes sporadiques
Le geste chirurgical dépend du siège de la tumeur.
Pour une tumeur gastrique de siège antral ou fundique :
Une gastrectomie atypique (wedge résection possible sans interruption de la continuité digestive) ou segmentaire (pour des raisons anatomiques) est indiquée. Le choix de la technique opératoire (cœlioscopie ou laparotomie) au-delà des critères de qualité oncologique classique (marge, reconstruction) repose aussi sur l’évaluation du risque de rupture peropératoire. Il est en effet essentiel d’éviter une effraction peropératoire de la tumeur qui péjore la survie globale.
La résection sous cœlioscopie ou robot-assistée est possible, mais elle doit être évitée en cas de volumineuse tumeur avec des remaniements importants kystiques, hémorragiques et/ou nécrotiques (Piessen, 2015) (accord d’experts).
Pour les tumeurs de siège pré-pylorique :
Une gastrectomie partielle des 4/5ème.
En cas de volumineuse tumeur, ou de tumeur proche du cardia, une gastrectomie totale peut être discutée ou en alternative une résection de la jonction œsogastrique avec interposition d’une anse intermédiaire de jéjunum pédiculée (intervention de Merendino).
Pour les tumeurs du grêle, une résection segmentaire sans curage est indiquée.
Pour les tumeurs duodénales, le type de chirurgie (conservatrice ou duodéno-pancréatectomie céphalique) dépend de la localisation et de la taille tumorale. Une chirurgie conservatrice du pancréas (duodénotomie) si elle est possible donne la même survie globale qu’une résection radicale (Duffaud, 2014) ; elle est donc à préférer (accord d’experts).
Pour les tumeurs du rectum et du côlon, une chirurgie réglée est classiquement recommandée. Néanmoins, une résection locale par voie transanale peut être discutée en RCP spécialisée en cas de GIST rectale de petite taille (Casali, 2022 ; Shu, 2020). Il est alors indispensable de passer au large de la tumeur, car les récidives sont fréquentes (accord d’experts).
Pour les tumeurs œsophagiennes, l’exérèse des GIST par énucléation sous thoracoscopie ou chirurgicale robot-assistée quand elle est possible peut être une alternative à l’œsophagectomie (Robb, 2015) (avis d’experts).
Lorsque la lésion est résécable, un traitement préopératoire par imatinib n’est pas indiqué (Casali, 2022 ; Von Mehren, 2014). En revanche, l’imatinib peut être indiqué après concertation pluridisciplinaire en centre spécialisé (et après analyse mutationnelle garantissent la sensibilité aux ITK) quand on estime qu’il peut modifier le geste opératoire en simplifiant la chirurgie ou en permettant une résection moins mutilante (préservation sphinctérienne pour le rectum par exemple) (cf. 12.3.6.1 imatinib en situation néo-adjuvante) (Shu, 2020). Ceci nécessite un suivi attentif afin de dépister précocement les 15 à 20 % de GIST d’emblée résistantes ou de moindre sensibilité à l’imatinib (KIT exon 9, etc.), et d’évaluer le risque hémorragique notamment lorsque le mode de diagnostic de la tumeur a été un saignement (accord d’experts).
12.3.3.3. Chirurgie des formes syndromiques
- Diade de Carney - Stratakis et triade de Carney :
Il s’agit de GIST multiples essentiellement gastriques, fréquemment associées à des métastases ganglionnaires et d’évolution indolente. L’équipe chirurgicale doit vérifier l’absence d’hypersécrétion de catécholamines qui pourrait faire prioriser la chirurgie d’un paragangliome. La stratégie est d’effectuer lorsque cela est possible une gastrectomie partielle ou en wedge afin d’avoir une meilleure qualité de vie. Un curage ganglionnaire sera systématiquement associé à la chirurgie du primitif.
L’argument contre la gastrectomie totale systématique d’emblée est que cela n’empêche pas les métastases hépatiques ultérieures et que la maladie est indolente (Kwak, 2023) (avis d’experts).
- GIST en lien avec une NF1 :
Dans un contexte de neurofibromatose de type 1, les GIST sont de localisation grêlique prédominante (86%), la plupart du temps KIT et PDGFRA sauvage. Elles sont très souvent multiples, la moitié étant découvertes sur des symptômes (21% de douleurs, 17% d’hémorragie, 5% d’occlusion) soit de façon fortuite dans le cadre du bilan d’une autre lésion liée à la maladie de Recklinghausen. La fréquence des complications augmente avec la taille (25% en dessous de 3 cm et 60% au-delà de 6 cm) (Hautefeuille, 2024).
Du fait de leur caractère moins évolutif, les lésions du grêle ayant un aspect de GIST et mesurant < 3 cm peuvent être surveillées. Par ailleurs, les GIST de moins de 3 cm opérées ne récidivent pas.
Le score RECKGIST permet d’évaluer le risque de récidive dans cette population spécifique (RECKGIST A : taille ≤ 30 mm quel que soit le nombre de mitoses, RECKGIST B si taille > 30 mm avec mitoses ≤ 5/5mm² ; RECKGIST C si taille > 30 mm et mitoses > 5/5 mm²) (Cuvelier, 2024). Au-dessus de 3 cm ou si la GIST est symptomatique, la chirurgie doit être discutée avec le patient (grade C).
Dans tous les cas, ces lésions doivent être surveillées. Le rythme de surveillance n’est pas codifié mais on pourrait proposer une TDM TAP à 6 mois puis à 18 mois puis tous les 2 ans si les lésions ne sont pas évolutives (accord d’experts).
Le risque de récidive est estimé à 15%, avec une survie sans récidive de 81% à 10 ans et 65% à 15 ans (Hautefeuille, 2024).
- GIST avec mutation germinale de KIT ou PDGFRA
GIST multiples et localisées au grêle ou à l’estomac : résection sélective des GIST symptomatiques ou à risque de complications et de celles qui progressent sous imatinib (lorsque la mutation était sensible) (avis d’experts).
12.3.4. Chirurgie des tumeurs localement avancées et/ou de résécabilité incertaine
Une exérèse large et mutilante n’est licite que si elle est complète avec une marge de tissus sain sur tout le pourtour de la tumeur visant à garantir l’exérèse complète et réduire le risque d’effraction tumorale. Cette attitude agressive est à moduler en fonction des organes concernés et du terrain. L’alternative d’un traitement néo-adjuvant par imatinib (si analyse mutationnelle en faveur d’une mutation prédictive de réponse) est judicieuse quand la résection semble importante ou incertaine en préopératoire, pour limiter le geste chirurgical initial et augmenter les chances de résection complète.
La chirurgie est alors envisagée quand la réponse RECIST maximale est observée généralement par CT TAP ou TEP-scanner au 18-FDG après 6 à 12 mois de traitement en moyenne (Casali, 2022). Sous imatinib préopératoire, donné habituellement à 400 mg/j, une imagerie CT ou TEP tous les 3 mois est proposée avec une indication chirurgicale qui sera possible une fois la tumeur stable (RECIST) sur 2 examens consécutifs (Choi, 2005). Il faut tenir compte du risque de complications sévères liées à la tumeur primitive (3% d’hémorragie, 3% de perforation, de rupture tumorale) sous imatinib, même s’il semble limité. Cette approche doit être réservée à des centres spécialisés (cf. 12.2.6.1) (accord d’experts).
12.3.5. Estimation du risque de récidive après résection à visée curative
L’estimation du risque de récidive est primordiale pour l’indication ou non d’un traitement adjuvant, et pour adapter la surveillance. L’imatinib est en effet le traitement adjuvant standard après résection d’une GIST à haut risque de récidive (grade A), et une option en cas de risque intermédiaire (accord d’experts) (cf. 12.2.6.2.) (Casali, 2022).
Une dizaine de classifications pour estimer le risque de récidive des GIST après résection microscopiquement complète (R0) ont été proposées. Elles présentent toutes des avantages et des inconvénients et sont basées sur des séries historiques rétrospectives avant l’avènement du traitement adjuvant.
Il en existe 2 grands types :
Celles classant les patients dans des groupes de risque de récidive estimé (NIH, NIH modifiée par Joensuu…). Les groupes ont été initialement définis de la manière arbitraire suivante : haut risque (>30 % de risque de récidive), risque intermédiaire (10-30 %), faible (<10 %) et très faible risque (0-2 %).
Celles estimant de manière chiffrée le risque de récidive (AFIP de Miettinen, nomogrammes, « contour maps » de Joensuu).
La première classification histo-pronostique du risque de récidive de Miettinen (tableau 1) était fondée sur la taille de la tumeur et l'index mitotique, a été établie en 2002 (Fletcher, 2002). L’index mitotique est le facteur pronostique de récidive le plus important avec la perforation le cas échéant. Le nombre de mitoses doit désormais être évalué sur 5 mm². Cela correspond, sur les microscopes actuels, à 20 à 25 champs au grossissement x 40 au lieu des « 50 champs à fort grossissement (HPF) » indiqués dans les anciennes publications. A noter que cette première ne prenait pas en compte la perforation qui est associée à un risque élevé de récidive (Miettinen, 2006).
De grandes séries rétrospectives ont permis de préciser ces données, intégrées dans la classification de Joensuu (tableau 2), mettant en évidence l’influence sur le risque de récidive du siège de la tumeur et de la perforation tumorale dans la cavité péritonéale ; cette dernière conférant un pronostic similaire à celui d’une GIST métastatique (Joensuu, 2008). Elle vise notamment à mieux scinder les GIST à risque intermédiaire et élevé, et intègre la perforation. Toutes les classifications sont utilisables en pratique, et aucune n’est parfaite. Les classifications AFIP (Armed Forces Institute of Pathology) de Miettinen (Miettinen, 2006) et NIH modifiée par Joensuu (Joensuu, 2008) sont les plus utilisées. Une classification TNM est disponible (UICC TNM8) et intègre les principaux facteurs pronostiques (Casali, 2022). Elle est peu utilisée en pratique. Enfin, d'autres paramètres topographiques, histologiques, immunohistochimiques et moléculaires impactent le risque de récidive mais ne sont pas encore pris en compte dans les classifications.
Tableau 2
Estimation du risque de récidive ou de décès lié à la maladie dans les GIST localisées réséquées dans des groupes définis par la taille,
l’index mitotique et le siège de la tumeur d’après Miettinen (Miettinen, 2006).
| Index mitotique ** | Diamètre maximal de la tumeur (cm) | GIST gastrique | GIST jéjuno-iléale | GIST duodénale | GIST rectale |
| ≤5 | ≤2 | 0 | 0 | 0 | 0 |
| ≤5 | >2 -5 | 1,9 % (très faible) | 4,3 % (faible) | 8,3 % (faible) | 8,5 % (faible) |
| ≤5 | >5 -10 | 3,6 % (faible) | 24 % (intermédiaire) | -* | -* |
| ≤5 | >10 | 12 % (intermédiaire) | 52 % (élevé) | 34 % (élevé) | 57 % (élevé) |
| >5 | ≤2 | 0 | 50 % (élevé) | -* | 54 % (élevé) |
| >5 | >2 -5 | 16 % (intermédiaire) | 73 % (élevé) | 50 % (élevé) | 52 % (élevé) |
| >5 | >5 -10 | 55 % (élevé) | 85 % (élevé) | -* | -* |
| >5 | >10 | 86 % (élevé) | 90 % (élevé) | 86% (élevé) | 71 % (élevé) |
* nombre de patients insuffisant pour l’estimation
** l’index mitotique est évalué par Miettinen sur une surface globale de 5 mm2, estimation des 50 champs à fort grossissement classiques afin de limiter la variabilité en fonction des microscopes (cela correspond en effet à seulement 20-25 champs à fort grossissement sur des microscopes récents).
Tableau 3
Estimation du risque de récidive dans les GIST localisées réséquées dans la classification de Joensuu (Joensuu, 2008)
| Risque de rechute | Taille | Index mitotique | Localisation |
| Très faible | ≤ 2 cm | ≤ 5 | Indifférente |
| Faible | >2-5 cm | ≤ 5 | Indifférente |
| Intermédiaire | ≤ 5 cm > 5-10 cm | 6-10 ≤ 5 | Gastrique Gastrique |
| Élevé | Indifférente > 10 cm Indifférente > 5 cm ≤ 5 cm > 5-10 cm | Indifférent Indifférent > 10 > 5 > 5 ≤ 5 | Rupture tumorale Indifférente Indifférente Indifférente Non gastrique Non gastrique |
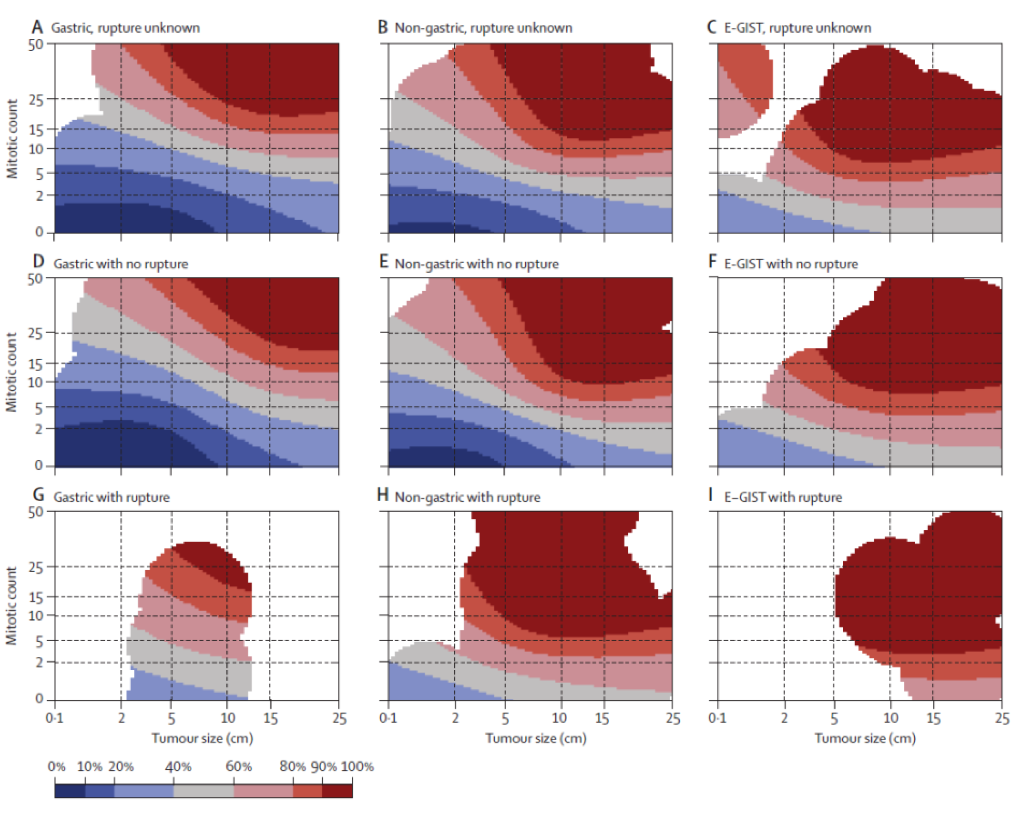
L’index mitotique et la taille étant des variables continues, les limites indiquées dans ces classifications doivent être interprétées judicieusement. Des « contour maps » pronostiques (cartes d’isolignes c’est-à-dire de répartition de même valeur du risque de récidive) qui incorporent l’index mitotique et la taille comme des variables continues ont été définies par Joensuu et al. à partir de séries de patients n’ayant pas reçu de traitement adjuvant (Joensuu, 2008).
Figure 2
Estimation du risque de récidive dans les GIST localisées réséquées selon les « contour maps » de Joensuu (Joensuu, 2012)
12.3.5.2. Facteurs de risques génomiques
Le génotype est un outil complémentaire pour évaluer le risque de récidive (Joensuu, 2015). La relation entre génotype et risque de récidive est complexe à analyser pour plusieurs motifs. D’une part, parce qu’il existe une grande variété de mutations possibles au niveau de l’exon 11 de KIT. Dans une étude 138 mutations différentes de KIT et PDGFRA étaient présentes chez 492 patients (Emile, 2012). Dix mutations représentaient cependant 56% de l’ensemble des mutations permettant certaines corrélations génotype/pronostic. D’autre part, outre sa valeur pronostique, la mutation a également une valeur prédictive de réponse au traitement par imatinib. Ainsi les mutations de l’exon 11 de KIT sont les plus sensibles à l’imatinib, alors que la mutation PDGFRA p.D842V est une mutation de résistance primaire à l’imatinib (Cassier, 2012).
En pratique, les GIST avec mutation de KIT ont un risque de récidive supérieur à celles avec mutation de PDGFRA, notamment celles avec mutation de l’exon 9. Les GIST sans mutation KIT/PDGFRA ayant un risque intermédiaire entre ces 2 groupes. Parmi les mutations de l’exon 11 de KIT, les délétions ont un risque de récidive supérieur aux substitutions et les duplications (plus rares) ont un meilleur pronostic (Joensuu, 2015).
D’autres facteurs moléculaires sont à l’étude. Il a été montré que le niveau de réarrangement du génome tumoral a une valeur pronostique (Lartigue, 2015). Un index génomique corrélé au risque de récidive a été déterminé, et est évalué dans une étude française prospective randomisée pour le traitement adjuvant de GIST de risque intermédiaire de rechute, étude close récemment (Essai GI-GIST).
Concernant les biomarqueurs de sensibilité aux immunothérapies par inhibiteurs de checkpoint immuns (anti PD-1, anti CTLA-4, etc), les GIST sont des tumeurs malignes ‘’froides’’, peu sensibles aux inhibiteurs de checkpoint immuns du fait d’un taux de néoantigènes (tumor mutation burden) faible et d’un microenvironnement tumoral riche en macrophages M2 immunosuppresseurs (TAMs) (Roulleaux Dugag, 2021).
12.3.6. Traitement (néo-)adjuvant des GIST localisées
12.3.6.1. Imatinib en situation néo-adjuvante
Il n’y a pas d’étude randomisée ayant évalué la place et les modalités de prescription d’un traitement néoadjuvant par imatinib (Eisenberg, 2009). La question se pose surtout pour les GIST gastroduodénales de grande taille et du rectum. L’objectif est la préservation d’organe et l’augmentation du taux de résection R0.
La dose la plus étudiée est 400 mg/j, un traitement de l’ordre de 6 à 12 mois permet d’obtenir un taux de réponse objective maximal, de l’ordre de 60 à 80 %. Dans les séries où la durée du traitement néo-adjuvant dépasse 6 mois, le taux de résection R0 est plus élevé, supérieur à 80 %. La durée optimale n’étant pas fixée et dépendant probablement des caractéristiques tumorales (localisation, mutation), l’attitude proposée est de réaliser un scanner tous les 2-3 mois et d’opérer lorsque le volume tumoral est le plus faible, ou après une stabilité sur 2 imageries consécutives (Rutkowski, 2013) (accord d’experts).
La médiane de traitement par imatinib dans cette série était de 10 mois. Le délai de réponse maximal est de 15 mois ; il n’est donc pas utile de traiter au-delà de 15 mois en situation néo-adjuvante. Le traitement adjuvant ou son absence sont déterminés par les facteurs pronostiques de la biopsie préopératoire (mitoses) et de l’imagerie (siège, taille) préopératoire. Si effectué, le traitement adjuvant sera proposé pour une durée totale de 3 ans incluant la durée de traitement néoadjuvant.
En cas de GIST (notamment gastrique) avec mutation p.D842V de l’exon 18 de PDGFRA ou de GIST non KIT/PGFRA mutée, l’imatinib néoadjuvant n’est pas conseillé (avis d’experts). En cas de mutation de PDGFRA, nous ne disposons d’aucune donnée pour indiquer l’avapritinib en néoadjuvant (avis d’experts) ; l’ESMO retient cependant l’avapritinib en néoadjuvant comme une option possible (Casali, 2022).
12.3.6.2. Imatinib en situation adjuvante
On dispose des résultats de trois essais de phase III.
Dans l’essai multicentrique américain ACOSOG Z9001, 773 patients avaient une GIST localisée de taille supérieure ou égale à 3 cm et une résection complète dans les 14 à 70 jours précédant l’inclusion (Dematteo, 2009). Les patients étaient randomisés entre imatinib 400 mg/j et placebo pendant un an. Le critère principal de l’étude était la survie sans récidive. A un an la survie sans récidive était de 97,7 % dans le bras imatinib versus 82,3 % dans le bras placebo (p<0,0001). Il n’a pas été observé de bénéfice sur la survie globale. Les résultats de l’analyse en sous-groupes selon les classifications NIH de 2002 et AFIP de Miettinen du risque de récidive n’ont pas montré de bénéfice en survie sans récidive dans les groupes à faible et très faible risque. Une analyse secondaire de sous-groupes en fonction du génotype a été rapportée (18). Un bénéfice significatif en survie sans récidive était observé en cas de mutation de l’exon 11 de KIT (présente chez 346 patients), mais pas en cas de mutation de l’exon 9 (35 patients) ou d’absence de mutation détectée (64 patients), notamment en cas de neurofibromatose. En cas de mutation PDGFRA (28 patients), il existait un risque de récidive spontané très faible, et aucun bénéfice de l’imatinib en cas de mutation D842V de l’exon 18 (mais bénéfice significatif en cas d’autres mutations de PDFGRA).
La seconde étude européenne SSGXVIII a comparé 1 an contre 3 ans d’imatinib 400mg/j chez 400 patients ayant une GIST à haut risque de rechute (selon la classification de Fletcher de 2002 avec par conséquent certains patients à risque intermédiaire selon la classification AFIP), ou avec rupture tumorale pré ou peropératoire (Joensuu, 2012). L’imatinib devait être débuté dans les 3 mois qui suivaient l’intervention chirurgicale. A 3 ans, la survie sans récidive était de 87 % dans le bras 3 ans d’imatinib contre 60 % dans le bras 1 an. Avec un suivi médian de 54 mois, la survie sans récidive était de 66 % contre 48 % en faveur du traitement de 3 ans (p<0,0001). La survie globale était meilleure à 5 ans avec un taux de 92 % dans le bras 3 ans contre 82 % dans le bras 1 an (p=0,019). Les résultats actualisés avec un suivi médian de 10 ans confirment la supériorité du schéma adjuvant 3 ans qui se maintient en termes de survie sans récidive (p=0,003) et de survie globale : 79 % contre 65 % à 10 ans (p=0,004) (Joensuu, 2020). Une analyse de sous-groupes en fonction du génotype montrait là encore que les patients ayant une mutation de l’exon 11 de KIT, notamment les délétions qui sont le plus fréquentes des mutations, bénéficient le plus du traitement adjuvant.
La troisième étude, EORTC 62024 a comparé imatinib 400 mg/j pendant 2 ans et surveillance chez 908 patients ayant une GIST à risque élevé ou intermédiaire (Casali, 2015). Le critère principal était la survie sans échappement à l’imatinib (IFS). Si la survie sans récidive était significativement meilleure dans le bras imatinib, l’IFS à 5 ans était similaire (87% contre 84%). Dans les GIST à haut risque, il existait une tendance non significative à une meilleure IFS (79% contre 73%) (p = 0,11). Il n’existait pas de bénéfice en survie globale. Cette étude suggère un effet plus suspensif que curatif de l’imatinib en adjuvant.
Une étude de phase II américaine a évalué le risque de récidive chez 91 patients ayant une GIST à risque élevé de récidive (en fait environ 1/3 des malades avaient un risque intermédiaire) traités par imatinib en adjuvant pendant 5 ans. La survie sans récidive estimée à 5 ans était de 90% et à 8 ans de 80%. Dans la plupart des cas, les récidives survenaient chez des patients n’ayant pas une mutation de l’exon 11 de KIT (Raut, 2018). Les patients ayant une mutation de l’exon 11 de KIT (environ 60 % des patients) sont les plus à même de tirer profit du traitement adjuvant, notamment les mutations de type délétion ou délétion-insertion ou celles impliquant les codons 557-558.
Des essais randomisés testant une durée plus longue de traitement adjuvant (> 3 ans) sont en cours. Les résultats de l’essai IMADGIST (Blay, 2024) conduit en France comparant arrêt de l’imatinib après 3 ans vs 3 ans supplémentaires (soit 6 ans) chez les patients ayant un risque de récidive estimé supérieur ou égal à 35% selon la classification NCCN ont été publiées dernièrement et sont en faveur d’un traitement adjuvant prolongé de 6 ans devant un bénéfice significatif en DFS (objectif principal) : DFS à 4 ans de 87% vs 55% ; HR=0,4 [0,20-0,67] p=0,008).
Un essai de traitement adjuvant comparant les durées de 6 ans versus 10 ans est en projet (IMADGIST-10).
REFERENCES
Classification :
Les discordances entre les 2 classifications NIH et AFIP pour estimer le risque de récidive posent un problème d’indication thérapeutique en particulier pour les GIST gastriques de 5 à 10 cm avec moins de 5 mitoses.
L’utilisation de la classification proposée par Miettinen et al. (AFIP) (Miettinen, 2006) et celle proposée par Joensuu (Joensuu, 2008) sont à privilégier pour estimer le risque de récidive, en s’aidant éventuellement des contour maps (présence ou non d’une perforation) (Joensuu, 2012) (avis d’experts) (Casali, 2022).
Génotypage :
La détermination du génotype de la tumeur est recommandée avant la mise en route d’un traitement adjuvant (accord d’experts).
Indication du traitement adjuvant :
Le traitement adjuvant est recommandé pour les GIST à haut risque de récidive (grade A).
Les GIST ayant une mutation de PDGFRA de type D842V de l’exon 18 (environ 20 % des GIST de l’estomac), mutation de résistance primaire à l’imatinib, ne tirent pas de bénéfice du traitement adjuvant et ont un risque de récidive spontané très faible (grade C).
Le traitement adjuvant n’est pas recommandé en cas de GIST liée à une perte d’expression ou une mutation de la SDH (GIST « SDH déficiente ») ou dans le cadre d’une neurofibromatose de type 1 (accord d’experts).
Posologie du traitement adjuvant : Imatinib 400 mg/j (grade A).
La durée du traitement adjuvant par imatinib recommandée est de 6 ans dans les GIST à haut risque (supérieur ou égal à 35% selon la classification NCCN) de récidive (grade A).
Imatinib adjuvant 800 mg/j :
Le bénéfice du traitement adjuvant dans les GIST sans mutation de KIT/ou PDGFRA ou ayant une mutation de l’exon 9 de KIT est mal connu. Certains comme les recommandation ESMO (Casali, 2022), préconisent l’imatinib à 800 mg/j en adjuvant en cas de mutation de l’exon 9 de KIT par analogie à la situation métastatique (Callejo, 2021) malgré l’absence d’étude prospective et une étude rétrospective négative (Vincenzi, 2022), cette attitude n’est pas partagée par la plupart des experts français (avis d’experts).
En cas de GIST mutée pour un exon dit de moindre sensibilité comme sur l’exon 9 de KIT, la dose adjuvante reste 400 mg/j (accord d’experts).
OPTION
Dans les GIST à risque intermédiaire, l’analyse du terrain, le génotypage de la tumeur, l’information et l’avis du patient sont des éléments importants pour la décision thérapeutique. Le traitement adjuvant pour une durée de 3 ans est une option (avis d’experts) dans l’attente des résultats de l’étude GI-GIST.
Dans les GIST perforées, le risque de récidive sous forme de GISTomatose péritonéale est majeur. La durée optimale du traitement adjuvant n’est pas définie chez ces patients virtuellement métastatiques qui pourraient bénéficier d’un traitement jusqu’à progression (avis d’experts).
12.3.6.3. Traitement adjuvant par avapratinib des formes PDGFRA D842V mutées
Il n’y a à l’heure actuelle aucune indication de traitement adjuvant par avapratinib, ce d’autant plus que les GIST p.D842V sont de très bon pronostic (avis d’experts).
12.3.6.4. Radiothérapie/chimiothérapie (néo-)adjuvante des formes localisées
La radiothérapie n’a été étudiée en adjuvant que dans de petites séries de patients qui avaient des facteurs de mauvais pronostic (envahissement local, marges envahies, rupture tumorale), sans que son intérêt ait été démontré. Il n’existe pas d’éléments en faveur de la chimiothérapie cytotoxique en situation adjuvante (accord d’experts).
12.3.7. Surveillance d’une GIST localisée
Les récidives après chirurgie sont essentiellement hépatiques et/ou péritonéales voire pleurales en cas d’invasion diaphragmatique. L’index mitotique de la tumeur influence la rapidité de survenue d’une récidive. Pour les patients à haut risque de récidive, elles surviennent surtout dans les 3 ans qui suivent la fin du traitement adjuvant. La majorité des récidives surviennent dans les 5 ans suivant la chirurgie ou la fin du traitement adjuvant. Des récidives plus tardives sont possibles mais rares (10).
Il n’existe pas de données dans la littérature permettant de valider un protocole précis de surveillance. Les protocoles de surveillance proposés correspondent donc à des avis d’experts (annexe 3).
L’exposition aux rayonnements ionisants et ses risques à long terme devant être pris en compte, d’autant plus que le patient est jeune et que la GIST a un risque de récidive bas, l’IRM abdominale est une alternative au scanner (Casali, 2022).
REFERENCES
- Examen clinique et TDM TAP injecté ou IRM abdominale (accord d’experts).
- Pas de rythme de référence établi par un essai, à discuter selon le risque de récidive et le terrain - Annexe 4.
OPTIONS (avis d’experts) (annexe 4)
- Tumeurs à risque élevé sous imatinib adjuvant : à 3 mois la 1ère année puis tous les 6 mois pendant 5 ans.
- Tumeurs à risque élevé après 6 ans d’imatinib adjuvant : tous les 3 mois pendant 2 ans puis tous les 6 mois pendant 3 ans, puis annuels pendant 5 ans.
- Tumeurs à risque intermédiaire (sans imatinib adjuvant) : tous les 3 à 6 mois pendant 3 ans, puis tous les 6 mois jusqu’à 5 ans, puis annuels jusqu’à 10 ans.
- Tumeurs à faible risque : à 6 mois puis tous les 12 mois pendant 5 ans.
- Tumeurs à très faible risque : pas de surveillance systématique.
12.4. Prise en charge des GIST avancées : inopérables ou métastatiques
Le réseau de référence clinique NETSARC (https://netsarc.sarcomabcb.org) labellisé par l'INCa comporte 25 centres spécialisés répartis sur tout le territoire national qui assurent la prise en charge de sarcomes des tissus mous et osseux, des GIST et des tumeurs desmoïdes. Il est recommandé de présenter les dossiers de GIST à une RCP spécialisée de ces centres.
Les métastases sont en général d’abord péritonéales et hépatiques puis plus tardivement pulmonaires, osseuses, musculaires, etc.
L’imatinib, un inhibiteur de tyrosine-kinases (TKI) dont KIT et PDGFRA, est le traitement de première ligne hors mutation PDGFRA p.D842V (cf 12.4.6.). La chirurgie n’est pas recommandée initialement au stade métastatique, mais peut être discutée en seconde intention sous traitement médical (Casali, 2022 ; Von Mehren, 2022).
12.4.1. Place de la chirurgie et des traitements locaux
La place de la chirurgie et des traitements locaux dans les GIST métastatiques reste discutée.
Avant traitement médical, l’exérèse de la tumeur primitive, si elle n’a pas eu lieu précédemment, n’est indiquée qu’en cas de symptômes majeurs (occlusion, hémorragie, perforation). Elle peut être discutée de manière multidisciplinaire en tenant compte du terrain et de l'importance du geste chirurgical avant le traitement par imatinib afin de prévenir la survenue de complications locales (avis d’experts).
LLe risque de perforation ou d’hémorragie de la tumeur primitive sous imatinib n’est pas connu précisément et est probablement limité (2,7 % d’hémorragie tumorale et 2,7 % de perforation dans une étude, pas de perforation dans deux autres études (Blanke, 2008 ; Demetri, 2009 ; Verweij, 2004).
Aucun bénéfice de la chirurgie n’a été démontré en cas de progression sous ITK qui est une indication de nouvelle ligne thérapeutique (accord d’experts).
Sous imatinib, chez les patients répondeurs ou stables et potentiellement accessibles à une résection complète, la place de la chirurgie d'exérèse ou d’un traitement local (thermo-ablation, radiothérapie stéréotaxique, cryo-ablation) de métastases reste à préciser (Casali, 2022 ; Von Mehren, 2022). Sa faisabilité a été démontrée, mais son bénéfice en termes de survie n'est pas établi. Deux essais randomisés (poursuite de l’imatinib versus chirurgie + poursuite de l’imatinib) ont été suspendus du fait d’un recrutement trop lent (27Du, 2014). Toute chirurgie dans ce cadre doit être discutée en réunion pluridisciplinaire (RCP), car le traitement par imatinib est le traitement de référence (Demetri, 2002) (accord d’experts). L’imatinib peut être arrêté la veille de la chirurgie et repris dès que le transit le permet.
Après imatinib ou ITK, chez les patients répondeurs ou stables, l’exérèse de volumineuses masses nécrotiques, symptomatiques, et dont le risque de rupture semble important est préférable à une chirurgie en urgence pour complication (accord d’experts). Une telle chirurgie doit être discutée en réunion de concertation pluridisciplinaire (RCP) spécialisée et doit être réalisée par une équipe entrainée.
Une exérèse ou une destruction de métastases peut être envisagée en cas de progression focale de la maladie sous imatinib pour retarder l’évolution générale de la maladie et l’introduction d’une 2ème ligne. Ce traitement n'a pas d’intérêt en cas de progression diffuse sous traitement médical (accord d’experts) (Casali, 2022).
En cas de découverte peropératoire d’une maladie métastatique lors de la résection de la tumeur primitive, il n’a pas été démontré de bénéfice d’une chirurgie de cytoréduction initiale des métastases. La résection du primitif associée à un traitement médical de phase avancée (cf. 12.4.3.) est indiquée en particulier si forme symptomatique ou à risques de complication. Après résection initiale macroscopiquement complète d’une maladie métastatique sans traitement médical préalable, un traitement adjuvant par imatinib est justifié (sauf en cas de mutation résistante à l’imatinib, cf. infra). Cette situation n’ayant pas fait l’objet d’essais spécifiques, sa durée (comme un traitement adjuvant ou jusqu’à progression) doit être discutée en RCP (avis d’experts) (Casali, 2022).
Après exérèse de métastases chez un patient en cours de traitement, la poursuite de l’imatinib est indispensable (accord d’experts) (Casali, 2022).
12.4.2. Place de la chimiothérapie et de la radiothérapie
Les chimiothérapies cytotoxiques ne sont pas efficaces dans les GIST (20De Pas, 2003). Des données montrent une certaine sensibilité des GIST non KIT, non PDGFRA, SDH déficiente et MGMT hyperméthylées au témozolomide (Giger, 2023).
La radiothérapie n’a été utilisée que ponctuellement, le plus souvent à visée symptomatique, pour des tumeurs fixées, responsables de douleurs ou hémorragiques, ou des métastases osseuses. Une série (phase II sur 25 patients) suggère qu’elle peut permettre dans certains cas des stabilisations de lésions abdominales ou hépatiques évolutives sous ITK (Joensuu, 2016).
12.4.3. Traitements médicaux en situation avancée
Préambule
Les traitements oraux en oncologie ayant un index thérapeutique très étroit, une attention particulière doit être portée aux potentielles interactions médicamenteuses. Les inhibiteurs de la pompe à protons semblent impacter négativement la survie de certains patients, ces traitements ayant un métabolisme commun au niveau des cytochromes hépatiques ou pouvant en modifier l’absorption. Si possible, les IPP (surtout avec le sunitinib) doivent donc être arrêtés (grade C).
Une cohorte prospective non randomisée, l’essai PRODIGE 92 - UCGI TARGET MONITO DIG est ouverte aux inclusions, avec notamment une cohorte GIST pour le sunitinib et le regorafenib avec mesure des concentrations plasmatiques en début de traitement, à progression et en cas d’effets indésirable sévère (PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG).
12.4.3.1. Imatinib
L’imatinib est le traitement de référence de première ligne des GIST localement avancées ou métastatiques (grade A), ayant transformé le pronostic de ces cancers chimiorésistants, avec une survie sans progression dans l’étude princeps de 20 à 33 mois (Blay, 2024 ; Casali, 2017 ; Demetri, 2002 ; Patrikidou, 2016).
En règle générale, ou lorsque le génotype de la tumeur n’est pas connu, la dose standard d’imatinib est de 400 mg/j, un comprimé en une prise au milieu d’un repas (grade A).
Le génotypage des tumeurs est recommandé (accord d’experts) (Casali, 2022 ; Von Mehren, 2014). Les GIST avec mutation de l’exon 11 de KIT sont les plus sensibles à l’imatinib. La posologie de 800 mg/j d’emblée est à privilégier en cas de GIST avec une mutation de l’exon 9 (grade C). Une méta-analyse de 2 essais de phase III a montré que les patients ayant une mutation de KIT sur l'exon 9 (10 % environ) avaient une survie sans progression augmentée en cas de traitement par 800 mg/j d’emblée (19 mois vs 6 mois ; p = 0,017). La survie globale était supérieure, mais de manière non significative, en cas de traitement par 800 mg/j d’emblée (35 mois vs 28 mois ; p = 0,15) (MetaGIST, 2010). Cependant, l’échantillon était limité (n =91) et le crossover autorisé lors d’une progression à 400 mg/j.
Les GIST sans mutation de KIT ou PDGFRA ont globalement une sensibilité moindre à l’imatinib que les GIST avec mutation de l’exon 11 de KIT. Un avis en centre expert est indispensable pour les patients ayant une GIST SDH-déficiente. Par ailleurs, l’imatinib est peu, voire pas actif chez les patients ayant une mutation de l’exon 18 de PDGFRA de type D842V (13) et l’avapritinib, inhibiteur spécifique de cette mutation, est maintenent disponible dès la 1ère ligne dans cette indication (cf 12.4.6.2.).
Il est recommandé de poursuivre le traitement par imatinib jusqu'à progression, intolérance ou refus du patient. Une augmentation de dose d’imatinib de 400 mg/jour à 400 mg 2 fois par jour en particulier en cas de durée de contrôle tumoral initiale prolongée (> 12 mois) (avis d’experts) ou un changement d’inhibiteur de tyrosine-kinases doit être discuté en cas de progression (cf. infra). Il n’est pas recommandé de diminuer la dose en l’absence de toxicité majeure, du fait d’une majoration du risque de progression (accord d’experts).
Des effets secondaires surviennent chez la majorité des patients, mais le plus souvent d’intensité modérée et régressant au cours du traitement. La tolérance de l’imatinib est dose-dépendante. Les quatre effets secondaires les plus fréquents sont les œdèmes, l’asthénie, les crampes et les troubles digestifs. L’observance du traitement doit être surveillée à chaque consultation. La prise en charge précoce et efficace des effets secondaires est la clé de l’observance aux inhibiteurs de tyrosine-kinases.
Intérêt du dosage plasmatique de l’imatinib
Il peut être intéressant de doser l’imatinibémie en cas de progression, de doute sur une interaction médicamenteuse ou de toxicité.
De la même manière que dans la leucémie myéloïde chronique, un taux plasmatique d’imatinib résiduel (Cmin) inférieur à 1100 ng/mL est associé à une absence de contrôle tumoral. Par ailleurs, un taux supérieur à 3180 ng/mL est associé à une augmentation de la toxicité (Demetri, 2009). Les adaptations de doses doivent être faites selon les RPC du produit, et peuvent être modulées en fonction des dosages plasmatiques de l’ITK du fait d’importantes variations interindividuelles (Demetri, 2009).
La résistance au traitement peut être primaire (dans les 6 premiers mois) (< 10 %), ou secondaire (après 6 mois). Il faut, avant de conclure à une résistance, éliminer un problème d’observance ou d’interactions médicamenteuses susceptible de diminuer l’exposition à l’imatinib (notamment jus de pamplemousse, inhibiteurs de la pompe à protons, millepertuis, etc). On peut consulter par exemple à ce sujet le thésaurus en ligne des interactions médicamenteuses de l’ANSM : https://ansm.sante.fr/documents/reference/thesaurus-des-interactions-medicamenteuses-1.
Un dosage du taux plasmatique résiduel d’imatinib est recommandé dans ce cas pour vérifier un taux plasmatiques résiduel d’imatinib >1100ng/mL (accord d’experts).
En cas de résistance secondaire avérée, on distingue les résistances partielles (évolution au niveau d'une ou d'un nombre limité de métastases) et les résistances multifocales (sur plusieurs lésions). L'arrêt de l'imatinib sans mise en route d’un autre traitement par inhibiteur de tyrosine-kinase est à éviter car elle est associée à une poussée évolutive des clones tumoraux sensibles même chez des patients en progression, avec un risque de progression plus rapide et intense et d’altération de l’état général pouvant compromettre la ligne ultérieure (accord d’experts). En cas de progression focale, un traitement local associé à la poursuite de l’imatinib (à la même dose ou à dose augmentée à 800mg/jour si exon 11 avec et/ou sous-exposition plasmatique et bonne tolérance) se discute (avis d’experts).
En cas de progression multifocale, l'inclusion dans un essai thérapeutique ou une augmentation des doses d'imatinib à 800 mg (qui se discute surtout en cas d’exon 11 avec de taux plasmatique bas d’imatinib ou de GIST avec mutation de l’exon 9 de KIT traitée à dose standard) permettant une stabilisation transitoire chez 30 à 40 % des patients – ou encore un changement d’inhibiteur de tyrosine-kinase est indiqué (accord d’experts) (Casali, 2022). Une demande d’avis auprès d’une RCP NETSARC est recommandée afin d’optimiser les inclusions dans les essais : 1 essai industriel avec le ripretinib est actuellement disponible en 2ème ligne en France.
Le traitement doit être poursuivi même en cas de progression avérée et relayé sans fenêtre thérapeutique (wash out) jusqu’à l’introduction de l’inhibiteur de tyrosine-kinase de ligne ultérieure afin d’éviter une progression rapide et intense pouvant compromettre la ligne ultérieure (accord d’experts). Une fenêtre thérapeutique est parfois imposée en cas d’inclusion dans un essai thérapeutique.
12.4.3.2. Critères d’évaluation par imagerie et surveillance au cours d’un traitement par inhibiteur de tyrosine-kinase pour GIST avancée ou métastatique (Casali, 2022 ; Von Mehren, 2014)
La tomodensitométrie avec injection de produit de contraste est l'imagerie la plus utilisée dans l'évaluation de la réponse (accord d'experts). Il a été montré que les critères RECIST de réponse tumorale n’étaient pas complètement adaptés à l'évaluation de la réponse tumorale dans les GIST traitées par imatinib. En cas de réponse, la masse devient hypodense, nécrotique et la partie prenant le contraste ainsi que la vascularisation tumorale diminuent en quelques semaines. Ces modifications ne sont pas toujours associées à une diminution de taille de la tumeur qui est plus lente, et peut même dans certains cas augmenter initialement. La mesure de la densité tumorale, en unités Hounsfield, est nécessaire. La diminution de la vascularisation tumorale évaluée par exemple par scanner dynamique avec injection de produit de contraste, traduit aussi l'efficacité du traitement (accord d'experts). Une augmentation de taille pouvant traduire l'efficacité du traitement, une revue de l'imagerie dans une RCP spécialisée cancérologie doit être envisagée avant son arrêt.
Des critères d’évaluation tomodensitométriques appropriés aux GIST ont été proposés pour définir le contrôle de la maladie sous imatinib (Choi, 2005) : diminution de la taille (mesure unidimensionnelle) > 10 % et/ou diminution de la densité après injection (en unités Hounsfield) d’au moins 15 %. Ces critères (dits de CHOI) ou encore les critères RECIST modifiés ont d’excellentes sensibilité et spécificité pour différencier les bons des mauvais répondeurs mais sont difficiles à appliquer en pratique.
L’IRM abdominale est une alternative à la tomodensitométrie abdomino-pelvienne.
La progression tumorale en cas de maladie métastatique traitée par imatinib peut être localisée (par exemple porter sur 1 ou 2 métastases, apparition d'un " nodule dans la masse " nécrotique), ou être diffuse. Les progressions focales représentent environ 50 % des progressions. Les modifications de densité intra-lésionnelle tumorale précèdent en moyenne de quelques mois les augmentations de taille des métastases. En cas de doute sur une progression, une relecture en centre expert est justifiée.
La TEP au FDG a montré une sensibilité élevée dans la détection de la réponse tumorale précoce (à 1 mois). Elle est toutefois d’intérêt limité en pratique courante pour l’évaluation de la réponse tumorale en situation avancée (Choi, 2007).
L'échographie de contraste permet aussi une évaluation précoce de la réponse par évaluation de la perfusion intra-tumorale des métastases hépatiques. Des logiciels permettent désormais d’obtenir une évaluation quantitative. Malgré sa simplicité et des études multicentriques concluantes, sa diffusion en pratique reste limitée et du domaine de la recherche.
L'amélioration symptomatique, les réponses tomodensitométrique (taille et densité) et TEP (SUV max) sont toutes prédictives du contrôle tumoral par l'imatinib.
REFERENCES
- Examen clinique, hémogramme et biologie hépatique tous les mois pendant 3 mois puis tous les 3 mois
- TDM thoraco-abdomino-pelvienne injecté avec mesure de densité des lésions tous les 3 mois ou IRM abdominale (accord d'experts).
OPTIONS
- Échographie de contraste (pré-thérapeutique puis dès J7 ou J28, puis tous les 3 mois) (avis d'experts).
- TEP au FDG (pré-thérapeutique puis dès J7 ou J28) si besoin d’évaluation précoce de l’efficacité (avis d'experts).
12.4.3.3. Mutations de résistance secondaires à l’imatinib et sensibilité aux autres TKI
Les mutations de résistance secondaires à l’imatinib sont le plus souvent en lien avec des mutations de KIT et affectant la boucle d’activation (activation loop soit les exons 17-18, environ 2/3 des cas) ou le site de liaison à l’ATP (ATP binding pocket soit les exons 13-14, environ 1/3 des cas). Ces mutations secondaires concernent majoritairement les exons 13 et 14 (site de liaison à l’ADN) et 17 et 18 (boucle d’activation).
Ces mutations de résistance secondaire à l’imatinib montrent des sensibilités différentes aux inhibiteurs de kinase de 2ème ligne et plus. Les mutations des exons 17 et 18 (activation loop) sont ainsi peu sensibles au sunitinib (Corless, 2011 ; Corless, 2014). L’étude INTRIGUE ayant comparé en 2ème ligne le sunitinib au ripretinib n’a pas montré de bénéfice du ripretinib sur l’ensemble de la population de l’étude laissant au sunitinb seul la place en 2ème ligne et au ripretinib sa place en 4ème intention (Bauer, 2022).
Néanmoins, des analyses post-hoc sur ADN tumoral circulant à progression montre que dans le sous-groupe des GIST initialement mutées KIT exon 11, l’apparition d’une néo-mutation de l’exon 13-14 semblait favoriser le sunitinib (PFS de 15 mois pour le sunitinib contre 4 mois pour le ripretinib). A l’inverse, dans le sous-groupe des GIST avec néo-mutation sur l’exon 17 ou 18, c’est le riprétinib qui semble être supérieur (PFS de 1.5 mois pour le sunitinib contre 14.2 mois pour le ripretinib). Les autres sous-groupes moléculaires soit favorisaient le sunitinb soit ne montraient pas de différence de sensibilité cliniquement significative (Heinrich, 2024).
Ces résultats sont actuellement en cours de validation dans l’essai essai industriel INSIGHT (https://clinicaltrials.gov/study/NCT05734105) ouvert aux inclusions dans différents centres français.
12.4.3.4. Sunitinib (Sutent®)
Le sunitinib est un inhibiteur oral de tyrosine-kinase agissant sur plusieurs récepteurs tyrosine kinase transmembranaires (KIT, VEGF, PDGF).
Il s’agit du seul inhibiteur de tyrosine-kinase ayant une AMM en deuxième ligne.
Son efficacité a été démontrée par une étude de phase III multicentrique chez 312 patients avec une GIST métastatique ou non résécable ayant une résistance ou une intolérance à l’imatinib où il permettait une PFS de 6.8 mois (Demetri, 2006, Heinrich, 2008).
Les ITK après la première ligne entrainent plus souvent des stabilisations (42%) de la maladie que des réponses objectives radiologiques (27.5%). La posologie classique (AMM) est de 50 mg/j 4 semaines sur 6. Un traitement continu à la dose de 37,5 mg /j aurait une efficacité similaire dans les GIST avec une tolérance comparable voire meilleure d’après une étude de phase II non randomisée (George, 2009).
L’observance du traitement est essentielle, et le traitement doit être personnalisé (posologie, schéma intermittent ou continu) selon la tolérance. La prise en charge précoce et efficace des effets secondaires est là encore indispensable. Du fait d’importantes variations interindividuelles, les dosages plasmatiques du sunitinib peuvent être une aide à l’adaptation des doses. De par son impact sur la sécrétion acide gastrique, la gastrectomie peut diminuer les concentrations de sunitinib. De la même manière, les IPP diminuent la sunitinibémie et ont un impact négatif sur la survie sans progression (7.7 mois sans IPP vs 3 mois avec IPP) (Mir, 2018). Les interactions médicamenteuses sont donc à prendre en considération.
Le sunitinib peut être dosé. Avec un schéma d’administration intermittent à 50 mg, la concentration minimale (Cmin) attendue pour l’efficacité doit être > 50 ng/mL et la Cmin pour la toxicité est > 80-87 ng/mL. Avec un schéma continu à 37.5 mg/j, la Cmin d’efficacité est > 37 ng/mL et la Cmin de toxicité de 60-75 ng/mL (Van der Kleij, 2023).
L’essai français PRODIGE 92 - TARGET MONITO DIG (PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG), ouvert aux inclusions, vise à documenter les concentrations plasmatiques de différents TKI avec une cohorte GIST pour le sunitinib et le regorafenib.
Les formes KIT mutées sur les exons 11 + 17 ou 11+ 18 en ADN tumoral circulant (confer 12.4.3.3) en 2ème ligne post imatinib doivent faire discuter l’inclusion dans l’essai de phase 3 industriel comparant sunitinib et ripretinib spécifiquement dans cette sous population moléculaire, essai en cours de recrutement en France (https://clinicaltrials.gov/study/NCT05734105). L’inclusion dans cet essai doit être privilégiée à la proposition de ripretinib hors AMM en 2ème ligne (avis d’experts).
12.4.3.5. Regorafenib (Stivarga®)
Le regorafenib est un inhibiteur oral de tyrosine-kinase agissant sur plusieurs récepteurs tyrosine kinase transmembranaires (KIT, VEGF, PDGF).
Il s’agit du seul inhibiteur de tyrosine-kinase ayant une AMM en troisième ligne, en cas d’échec et/ou intolérance de l’imatinib et du sunitinib.
Son efficacité a été démontrée par une étude de phase III multicentrique contre placebo, chez patients avec une GIST métastatique ou non résécable, résistants ou intolérants à l’imatinib et au sunitinib: médiane de survie sans progression de 4.8 versus 0.9 mois (HR=0.27) (Demetri, 2013).
La dose préconisée est de 160 mg/j 3 semaines sur 4. Comme pour tous les TKI la prévention et la prise en charge des effets secondaires, l’adaptation des doses à la tolérance, et la personnalisation du traitement, sont essentiels pour l’observance. Même en l’absence de données spécifiques aux GIST, un schéma d’escalade de dose de 80 mg puis 120 et 160 mg/j peut être proposé comme dans le cancer colorectal (étude ReDOS) (avis d’experts).
12.4.3.6. Ripretinib (Qinlock®)
Le ripretinib a été évalué dans un essai randomisé de phase III contre placebo avec cross-over en 4ème ligne thérapeutique, l’essai INVICTUS (Blay, 2020). Cet essai montre une supériorité du ripretinib 150mg par jour en continu pour le taux de réponse (11% vs 0%) et pour la survie sans progression (médiane à 6.3 mois vs 1.0, HR=0.15).
Le médicament est maintenant remboursé depuis le 22 octobre 2024 en cas de GIST avancée ayant reçu au préalable au moins 3 TKI dont l’imatinib (grade A).
A noter que le ripretinib semble efficace quel que soit le sous-groupe de mutation secondaire acquise (exon 9, 11, 13, 17 et aussi pour les exons 11 + 13, 11 + 17).
La place du ripretinib en 2ème ligne post imatinib dans les formes mutées sur ADN tumoral circulant sur les exons 11 + 17 ou 18 de KIT doit être discutée en RCP spécialisée de façon à privilégier pour les patients concernés, l’inclusion dans l’essai industriel ouvert actuellement aux recrutements en France et cherchant à valider prospectivement la place du ripretinib en 2ème ligne dans cette population (https://clinicaltrials.gov/study/NCT05734105) (avis d’experts) (cf 12.3.3.3. et 12.3.3.4.).
A progression sous dose standard de 150mg par jour en continu, un doublement de la posologie à 150 mg x 2 par jour est possible (avis d’experts) permettant un gain cliniquement significatif de la survie globale dans l’essai INVICTUS au détriment d’une toxcité hématologique accrue (anémie essentiellement) (Zalcberg, 2021).
12.4.3.7. Avapritinib (Ayvakit®)
L’avapritinib a montré un taux de réponse de 91% et une médiane de survie sans progression proche de 34 mois pour les patients porteurs de mutations PDGFRA D842V inopérables ou métastatiques (Jones,2021). Le profil de tolérance montre outre de fréquents (>60% des patients) nausées, vomissements et anémie, une toxicité cognitive (>40% des patients) sous formes de troubles mnésiques, de confusion voire d’encéphalopathie. A noter également un risque faible (5%) d’hémorragie intracrânienne.
La prescription d’avapritinib devra être prise après proposition issue d’une RCP avec un centre de référence et de compétences dans la prise en charge des sarcomes (recommandation HAS). Il dispose d’une AMM européenne dans les GIST inopérables ou métastatiques avec mutation PDGFRA D842V sans spécification de ligne thérapeutique (grade B).
Il est disponible en pharmacie d’officine depuis le 4 avril 2024.
12.4.3.8. Inhibiteurs de NTRK
Le larotrectinib (Vitrakvi®) est efficace en cas de fusion du gène NTRK à rechercher pour les GIST sans mutation KIT et PDGFRA (Drilon, 2018 ; Marchio, 2019). Il a obtenu une AMM conditionnelle de l’EMA en 2019 dans les tumeurs solides impliquant l’un des gènes NTRK sans autre alternative thérapeutique Ce médicament n’est actuellement remboursé en France que chez les patients pédiatriques ayant un fibrosarcome infantile ou un autre sarcome des tissus mous avec une fusion du gène NTRK. Un autre médicament, l’entrectinib (Rozlytrek®), appartenant à cette même classe thérapeutique des anti-NTRK a obtenu une AMM conditionnelle de l’EMA en septembre 2020. Pour mémoire, ces altérations de NTRK sont très rares dans les GIST (0.1 -0.3% des cas).
12.4.3.9. Anti-BRAF, nouveaux TKI et immunothérapies
La mutation BRAF V600E prédictive de réponse aux anti-BRAF sont très rares dans les GIST, leur incidence est estimée autour de 1%. L’identification de cette mutation légitime le recours aux anti-BRAF (dabrafenib, encorafenib, vemurafenib) par analogie aux mélanomes avancés (Calderillo-Ruiz, 2024) (avis d’experts).
De nouvelles molécules prometteuses font l’objet d’études notamment après échappement à l’imatinib et au sunitinib dans les GIST métastatiques, et peuvent être accessibles par l’inclusion dans les essais dédiés. Il s’agit principalement d’inhibiteurs multikinases ayant une efficacité sur des mutations de résistance secondaire à l’imatinib de KIT (exons 13-14/17-18) et PDGFRA comme le NB003 (Chi, 2023) ou des inhibiteurs de HSP90 (Naito, 2023). Certains ont une efficacité potentielle en cas de mutation PDGFRA D842V (crenolanib). Des inhibiteurs de VEGFR2 (vandetanib) et des inhibiteurs de points de contrôle immunitaires (ICI) sont en cours d’évaluation. Les ICI n’ont pas montré d’intérêt significatif en monothérapie et sont évalués actuellement en combinaison avec les TKI. L’atezolizumab en combinaison avec l’imatinib est en cours d’évaluation versus imatinib chez des patients en échec thérapeutique dans l’essai ATEZOGIST du Groupe Sarcome Français (recrutement ouvert). L’association du regorafenib avec l’avelumab dans l’essai multi-tumeur REGOMUNE (https://classic.clinicaltrials.gov/ct2/show/NCT03475953) montre des résultats modestes en 3ème ligne, avec une PFS à 6 mois de 57% dans le bras GIST gastrique, supérieure à la PFS du bras GIST du grêle qui était à 17% (Cousin, 2023).
12.4.3.10 Rechallenge par imatinib et autres TKI de nème ligne
Le rechallenge par imatinib est une option thérapeutique envisageable après recours à tous les TKI validés par des études randomisées en 1ère (imatinib), 2ème (sunitinib), 3ème (régorafenib) et 4ème (ripretinib) ligne (hors GIST mutées PDGFRA p.D842V qui imposent le recours préalable à l’avapritinib) telle qu’attesté par une étude randomisée de phase 3 de 2013 montrant une médiane de PFS doublée versus placebo (Kang, 2013) (accord d’experts).
Parmi les inhibiteurs multikinases ayant montré une certaine efficacité dans des phases II mais non développés en phase III dans les GIST et n’ayant pas d’AMM on peut citer : le sorafenib (Nexavar®), le pazopanib (Votrient®), le nilotinib (Tassigna®) (pas de supériorité sur l’imatinib dans les GIST lors de l’analyse intermédiaire d’une phase III en première ligne et moindre efficacité en cas de mutation de l’exon 9 de KIT), le dasatinib (Sprycel®), le cabozantinib (Cabometyx®) et dernièrement, le lenvatinib (Lenvima® / Kisplyx®) sur des données de phase 2 randomisée versus placebo de l’essai français LENVAGIST (Le Cesne, 2024).
12.4.3.11. Formes ‘’super wild type’’ (non KIT non PDGFRA non BRAF non RAS mutées et non NTRK altérées) et formes syndromiques
L’efficacité des inhibiteurs de tyrosine-kinase susmentionnés et utilisés dans les GIST avec mutation oncogénique identifiée (KIT, PDGFRA) est très faible (stabilité au mieux) voire inexistante dans les formes non KIT non PDGFRA non BRAF non RAS mutées et non NTRK altérées. De même, dans les formes syndromiques. De plus, ces rares GIST ont une évolution indolente avec un pronostic favorable au long cours.
Ces 2 caractéristiques font de ces GIST des indications privilégiées d’inclusion dans des essais thérapeutiques. En l’absence d’essai disponible, la succession classique de ligne des GIST KIT mutée est à privilégier après validation en RCP spécialisée : imatinib – sunitinib – regorafenib – ripretinib (avis d’experts).
Les chimiothérapies cytotoxiques ne sont pas efficaces dans les GIST. Des données montrent cependant une certaine sensibilité des GIST non KIT, non PDGFRA, SDH déficiente et MGMT hyperméthylées au témozolomide, avec des stabilités prolongées dans ces sous types moléculaires rares (Giger, 2023).
12.5. Indications thérapeutiques
Toutes les décisions thérapeutiques concernant une GIST doivent faire l’objet d’une RCP. Un avis auprès d’un centre régional de référence dans la prise en charge des sarcomes et tumeurs conjonctives (réseau NETSARC, https://netsarc.sarcomabcb.org) est recommandé pour privilégier l’inclusion dans les essais et devrait être systématique. La majorité des recommandations émanent de l’avis d’experts. Des algorithmes schématisant les principales indications thérapeutiques figurent en annexes (annexes 3 et 4).
12.5.1. GIST résécable non métastatique, résection R0
REFERENCES
Chirurgie d'exérèse R0 (grade A).
Imatinib 400 mg/j en adjuvant pendant 6 ans si GIST à haut risque de récidive (grade A).
Pas de traitement adjuvant pour les GIST avec mutation PDGFRA de type p.D842V (grade B) ou mutation NF1 ou GIST « SDH déficiente » (accord d’experts).
Pas de traitement adjuvant pour les GIST en lien avec une NF1 (grade C), sauf pour les rares GIST mutées KIT ou PDGFRA (cf. indications supra)
OPTIONS
Imatinib en adjuvant pendant 3 ans si GIST à risque intermédiaire de récidive (avis d’experts) ; tenir compte notamment du terrain, du statut mutationnel de la tumeur (exon 11 de KIT ou non) et du souhait du patient.
GIST perforée : pas d’accord sur la durée du traitement adjuvant : 6 ans (résultats IMADGIST ; 2 autres essais évaluant durée > 3 ans en cours) (accord d’experts).
GIST mutées KIT exon 9 : imatinib 400mg/ jour d’emblée (avis d’experts).
Cas particulier des petites GIST (accord d’experts) :
• Si GIST de l’estomac < 2 cm de diamètre : surveillance ou résection chirurgicale. Si surveillance : écho-endoscopie (ou à défaut endoscopie) à 6 mois, 18 mois puis tous les 2 ans, à adapter en fonction du terrain et de la croissance éventuelle de la lésion.
• Si GIST du grêle, du rectum ou du duodénum : résection chirurgicale quelle que soit la taille (cf. chapitre 12.2.1.)
• Si GIST gastrique (< 2 ou 3 cm), la résection endoscopique par dissection sous-muqueuse type FTRD (« full-thickness resection device ») est une option dans un centre expert, éventuellement par une approche combinée avec la cœlioscopie (avis d’experts).
12.5.2. GIST résécable non métastatique, résection R1 ou R2
REFERENCES
- Pas de référence
OPTIONS
- Discuter une reprise chirurgicale surtout en cas de résection R2 car la valeur péjorative d’une résection R1 n’est pas clairement établie (avis d’experts). Si la séreuse est envahie ou s’il y a une effraction tumorale, le pronostic est surtout lié à l’essaimage péritonéal et non plus à la tranche de section viscérale et une reprise d’exérèse n’est donc pas justifiée. La reprise se discute lorsque que le patient est potentiellement curable par la chirurgie (séreuse non envahie, pas d’effraction tumorale et lésion à risque faible ou intermédiaire de récidive).
- Si une reprise n’est pas possible, que la résection est R2 (macroscopiquement incomplète) il faut discuter un traitement par imatinib et une reprise chirurgicale dans un deuxième temps (accord d’experts).
- Imatinib en adjuvant pendant 6 ans si GIST à risque élevé ou 3 ans si risque intermédiaire de récidive et résection R1 (accord d’experts).
- Imatinib en post-opératoire sans limitation de durée si résection R2 et pas de reprise chirurgicale possible (avis d’experts).
12.5.3. GIST résécable, mais survenant dans un contexte de prédisposition familiale
REFERENCES
- Pas de référence
OPTIONS (avis d’experts)
- Contexte de neurofibromatose de type 1 : les GIST sont souvent multiples sur le grêle, à ne pas confondre avec des métastases. En cas de petite GIST asymptomatique, la surveillance est possible. Indication opératoire en cas de complication (occlusion, douleur, hémorragie) ou en cas de GIST progressive ou de GIST > 2 cm. Le traitement adjuvant par imatinib n’est pas démontré et n’est pas recommandé. Contact recommandé avec les centres de compétences régionaux de prise en charge de la neurofibromatose.
- Contexte de syndrome de Carney-Stratakis ou de perte d'expression de SDHB (GIST épithélioïdes gastriques du sujet d'âge < 30 ans). Des métastases ganglionnaires ont été rapportées dans 30% des cas ce qui fait discuter un curage ganglionnaire (même en cas de mise en évidence en post-opératoire de la lésion primitive).
- Contexte de mutation germinale de l'exon 13 de KIT : un traitement prolongé par imatinib peut être proposé en cas de GIST symptomatique, de plus de 3 cm et/ou à croissance rapide.
12.5.4. GIST de résécabilité douteuse ou chirurgie mutilante (œsophage ou rectum en particulier)
REFERENCES
- Pas de référence.
OPTIONS
- Discuter un traitement néo-adjuvant par imatinib 400 mg/j en vérifiant qu’il n’y ait pas de résistance primaire à l’imatinib (grade C).
- Résection chirurgicale secondaire dans un centre spécialisé à discuter au nadir de la réponse objective évaluée par TDM (accord d'experts).
- Imatinib adjuvant pour une durée totale de 6 ans si haut risque (grade A) en prenant en compte la taille initiale et le nombre de mitose le plus élevé.
- GIST mutées PDGFRA p.D842V : avapritinib à proposer en considérant la lésion inopérable (AMM) et en interrogeant une chirurgie secondaire si réponse objective (accord d’experts).
ESSAIS CLINIQUES
Pas d’essai néo-adjuvant actuellement ouvert en France.
12.5.5. GIST non résécable, non métastatique
REFERENCES
- Traitement par imatinib 400 mg/j (accord d'experts).
- Résection chirurgicale secondaire dans un centre spécialisé à discuter au maximum de la réponse objective, après 6 à 12 mois d’imatinib (accord d'experts).
OPTIONS
- Traitement par imatinib 800 mg/j d’emblée si mutation de l’exon 9 de KIT connue (avis d’experts).
- GIST avec mutation PDGFRA D842V inopérables ou métastatiques (pas de spécification de ligne thérapeutique) : avapritinib (cf. 12.4.3.7) (avis d’experts).
ESSAIS CLINIQUES
Pas d’essai néo-adjuvant actuellement ouvert en France
12.5.6. GIST métastatique
REFERENCES
- Traitement par imatinib 400 mg/j (grade A).
- Exérèse de la tumeur primitive à discuter si risque de complication évaluée comme important (en particulier rupture d’une volumineuse tumeur liquéfiée) selon l’importance du geste opératoire (accord d'experts).
- Traitement par imatinib 800 mg/j d’emblée si mutation de l’exon 9 de KIT (accord d’experts).
- Dosage plasmatique de l’imatinib pour adaptation posologique si inefficacité ou toxicité
- GIST avec mutation PDGFRA p.D842V inopérables ou métastatiques (pas de spécification de ligne thérapeutique) : Avapritinib (AYVAKYT®) (grade B).
OPTIONS
- Si réponse ou stabilité et résection R0 potentiellement réalisable : résection et/ou radiofréquence des métastases après traitement par imatinib (procédure expérimentale), et poursuite de l’imatinib en post-opératoire (avis d’experts).
- Exérèse de métastases nécrotiques sous imatinib si risque de complication, en particulier de rupture de masses liquéfiées (accord d'experts).
- Cas particulier 1 : résection initiale (avant tout traitement par imatinib) complète d’une maladie métastatique limitée dans le même temps que la tumeur primitive : traitement adjuvant par imatinib si pas de mutation PDGFRA D842V pour une durée indéterminée (3 ans ou plus) à discuter en RCP (avis d'experts).
- Cas particulier 2 : en cas de GIST sans mutation de KIT ou PDGFRA retrouvée, d’évolution lente, recours à la destruction ou à l’exérèse des métastases plus fréquente (avis d'experts).
- Cas particulier 3 : En cas de GIST « SDH déficiente », contact avec un centre NETSARC pour avis (accord d'experts).
ESSAI CLINIQUE
- Etude GIST-TEN : étude de Phase II, prospective, randomisée, multicentrique, en ouvert, évaluant l'intérêt d'interrompre ou de maintenir l'imatinib chez les patients ayant une GIST localement avancée/métastatique après 10 ans de traitement. Coordonnateur : Pr JY Blay, Centre Léon Bérard, Lyon https://clinicaltrials.gov/ct2/show/NCT05009927
12.5.7. Progression sous imatinib 400 mg/j
REFERENCES (accord d'experts)
Avis auprès d’une RCP du réseau NETSARC.
Pas d’arrêt immédiat de l’imatinib avant la décision thérapeutique.
Authentifier la progression radiologique (critères CHOI ou RECIST modifiés).
Vérifier l’observance du traitement.
Eliminer des interactions médicamenteuses et si possible contrôler l’exposition au traitement : dosage plasmatique du taux d’imatinib >1100 ng/mL (grade C).
Augmentation de l’imatinib à 800 mg/j si la tolérance à 400 mg/j le permet, surtout si sous-exposition à l’imatinib ou mutation de l’exon 9 de KIT traitée à 400 mg/j (avis d'experts).
Changement d’ITK : sunitinib 50 mg/j 4 semaines sur 6 (grade A), si possible dans le cadre de l’essai PRODIGE 92 - UCGI TARGET MONITO DIG (PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG).
Ou essais industriels de 2ème ligne avec le ripretinib si mutation exon 11 + 17 ou 18 en ctDNA (https://clinicaltrials.gov/study/NCT05734105).
• GIST avec mutation PDGFRA p.D842V : avapritinib si non administré lors de la première ligne (grade B).
OPTIONS
Identification mutation de résistance à l’imatinib sur ADN circulant exclusivement pour discuter en RCP spécialisée la place du ripretinib en 2ème ligne post imatinib dans les formes mutées sur les exons 11 + 17 ou 18 de KIT ; cette situation doit faire privilégier l’inclusion dans l’essai industriel évaluant la place du ripretinib dans cette situation (accord d’experts).
Résection ou traitement ablatif voire embolisation des métastases si progression focale sous imatinib et discuter augmentation de l’imatinib à 800 mg/j (avis d’experts).
Sunitinib en continu à 37,5 mg/j plutôt qu’à 50 mg/j 4 semaines sur 6 (avis d’experts).
Dosage plasmatique de l’ITK (imatinib, sunitinib) ; arrêt des IPP ou poursuite avec dosage du sunitinib pour s’assurer d’être en zone thérapeutique.
ESSAIS CLINIQUES
Prendre contact avec un centre du réseau NETSARC pour information sur les essais en phase de recrutement ou à venir
PRODIGE-92 TARGET MONITO DIG (sunitinib) (PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG).Ou avec ripretinib si mutation exon 11 + 17 ou 18 en ctDNA (https://clinicaltrials.gov/study/NCT05734105)
12.5.8. Progression après imatinib et sunitinib
REFERENCES
- Regorafénib 160 mg/j, 3 semaines sur 4 (grade A), si possible dans le cadre de l’essai PRODIGE 92 - UCGI TARGET MONITO DIG (PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG).
Adaptation du traitement à la tolérance clinique, schéma d’escalade de dose pour augmenter la tolérance du traitement. Prévention et prise en charge précoce des effets secondaires. Vérification de l’observance.GIST avec mutation PDGFRA D842V : avapritinib si non administré dans une ligne antérieure (grade B).
ESSAIS CLINIQUES
- Prendre contact avec un centre du réseau NETSARC pour information sur les essais en phase de recrutement ou à venir
- Essai TARGET MONITODIG PRODIGE 92-UNICANCER-UCGI 42 TARGET-MONITODIG
Essai REGOMUNE (cohorte GIST) : EEssai de phase I-II régorafénib +avelumab (anti PD-L1). Coordonnateur : Dr Cousin, Institut Bergonié, Bordeaux. https://classic.clinicaltrials.gov/ct2/show/NCT03475953 - Essai ATEZOGIST : atezolizumab en combinaison avec l’imatinib versus imatinib seul chez des patients en échec thérapeutique après imatinib et sunitinib. Coordonnateur : Dr Brahmi, Centre Léon Bérard, Lyon Synopsis ATEZOGIST.pdf
12.5.9. Traitement au-delà de la 3ème ligne et au-delà
REFERENCES
Ripretinib (QUINLOCK) (grade A) remboursé GIST inopérable ou métastatique chez les patients ayant déjà reçu au moins 3 lignes de traitement par ITK.
Au-delà du Ripretinib :
- Autres ITK en particulier le lenvatinib (grade C), en escalade de dose si patients fragiles.
- Rechallenge Imatinib idéalement dans le cadre de l’essai ATEZOGIST Synopsis ATEZOGIST.pdf (accord d’experts).GIST avec mutation PDGFRA D842V : avapritinib si non administré dans une ligne antérieure (accord d’experts).
OPTIONS
Si fusion du gène NTRK, larotrectinib (per os 200 mg/j) (non remboursé) (pour GIST sans mutation KIT et PDGFRA) sans aucune autre option thérapeutique satisfaisante (sélection par test immuno-histochimique confirmé par séquençage (NGS ARN) ou séquençage (NGS ARN) d’emblée) (grade C) (25).
Autres TKI testées en phase 2 dans les GIST, mais prescription hors AMM sous la responsabilité du médecin et après information du patient, si pas d’essai disponible, après validation en RCP NETSARC (avis d’experts).
ESSAIS CLINIQUES
Essai ATEZOGIST : atezolizumab en combinaison avec l’imatinib versus imatinib seul chez des patients en échec thérapeutique après imatinib et sunitinib. Coordonnateur : Dr Brahmi, Centre Léon Bérard, Lyon Synopsis ATEZOGIST.pdf
Essais de phase I si état général conservé (PS 0-1) : s’adresser à un centre d’essais précoces : www.e-cancer.fr/Professionnels-de-la-recherche/Recherche-clinique/Structuration-de-la-recherche-clinique/Les-CLIP2
12.6. Annexes : algorithmes de prise en charge
Annexe 1. Indications de la consultation d’oncogénétique et algorithme d’analyse génétique constitutionnelle en fonction de l’immunohistochimie SDH
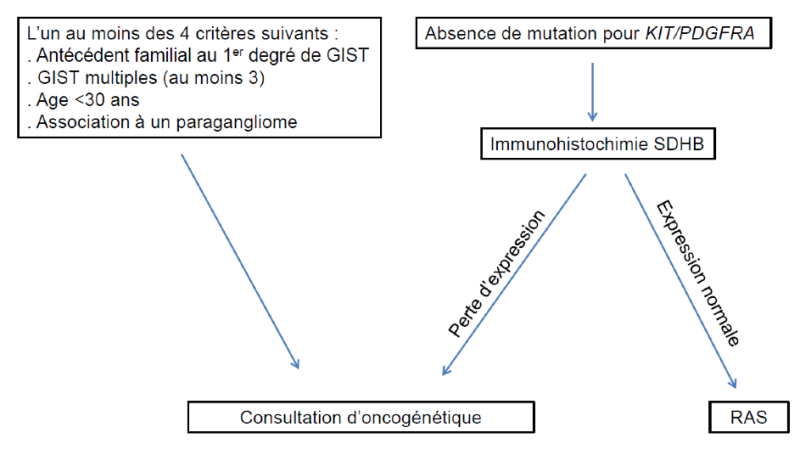
Annexe 2. Algorithme en cas de suspicion de GIST gastrique de petite taille (d’après réf 6.)
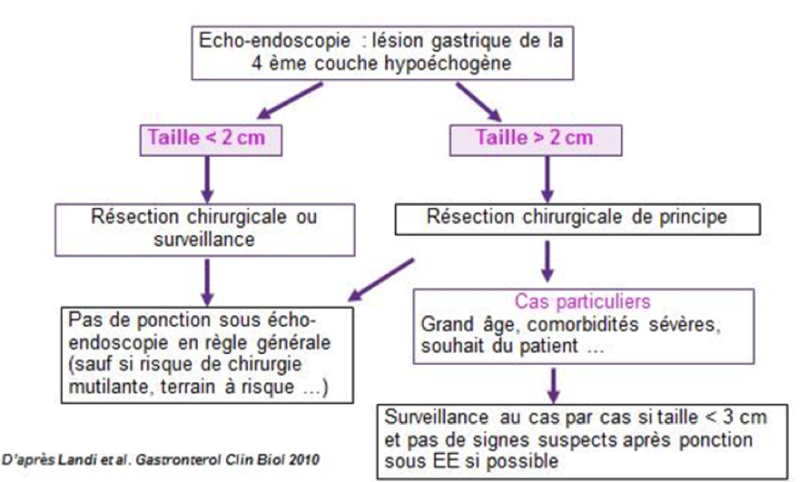
Annexe 3. GIST localisées : algorithme schématique de prise en charge et surveillance (d’après réf 1.)
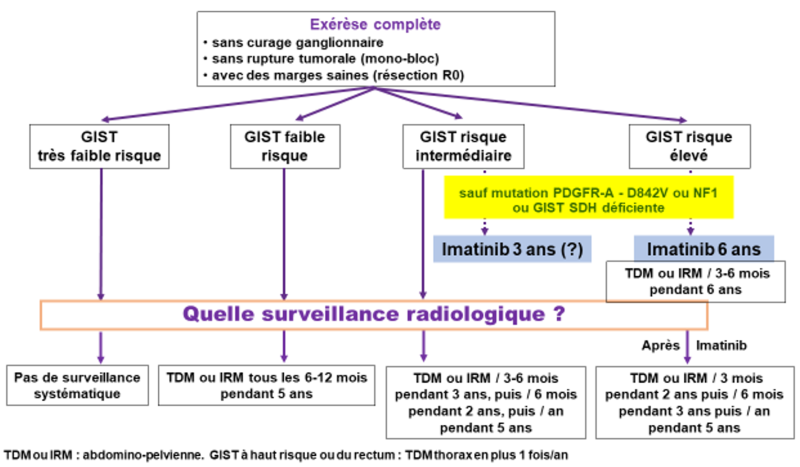
Annexe 4. GIST métastatiques ou localement avancées : algorithme de prise en charge (d’après réf 1.)
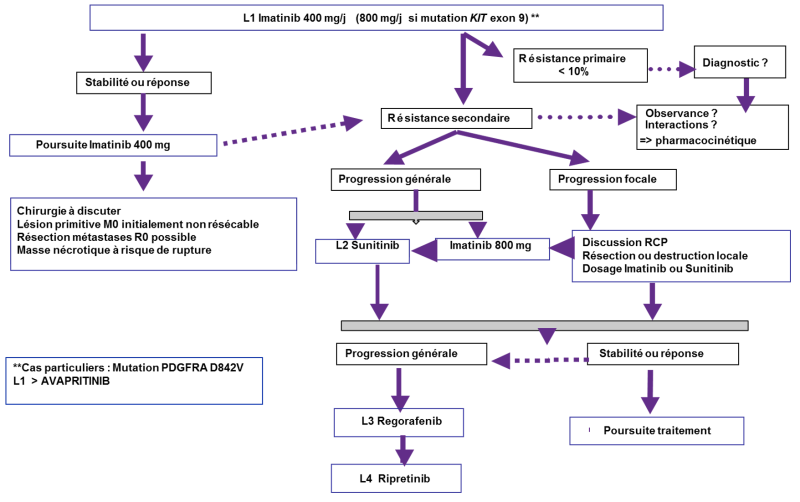
12.7. Références bibliographiques
Bachet JB, Landi B, Laurent-Puig P et al. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (GIST) and germline mutation of KIT exon13. Eur J Cancer 2013; 49:2531-41
Bauer S, Jones RL, Blay JY et al Ripretinib versus Sunitinib in Patients with Advanced GIST after Imatinib (INTRIGUE) : a Randomized, Open-label, Phase III Trial. J Clin Oncol 2022 ; 40(34) :3918-3928
Blanke CD, Rankin C, Demetri GD et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the KIT receptor tyrosine kinase: S0033. J Clin Oncol 2008; 26:626-32
Blay JY, Serrano C, Heinrich MC et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2020; 21:923-934.
Blay JY, Devin Q, Duffaud F et al. Discontinuation versus continuation of imatinib in patients with advanced gastrointestinal stromal tumours (BFR14): exploratory long-term follow-up of an open-label, multicentre, randomised, phase 3 trial. Lancet Oncol 2024; 25(9): 1163-1175
Blay JY, Schiffler C, Bouché O et al. A randomized study of 6 vs 3 years of adjuvant imatinib in patients with localized GIST at high risk of relapse. Ann Oncol 2024; in press doi.org/10.1016/j.annonc.2024.08.2343
Bouchet S, Poulette S, Titier K et al. Relationship between imatinib trough concentration and outcomes in the treatment of advanced gastrointestinal stromal tumours in a real-life setting. Eur J Cancer 2016; 57:31-8.
Calderillo-Ruiz G, Perez-Yepez EA, Garcia-Gamez MA et al. Genomic profiling in GIST : implications in clinical outcome and future challenges. Neoplasia 2024 ; 48 p. 100959
Callejo A, Faouzi S, Bouché O et al. Starting Imatinib at 400mg daily in Patients with GIST harboring KIT Exon 9 Mutations: a Retrospective, Multicenter Study. Target Onco 2021 vol 16(4) pp 485-492
Casali PG, Blay JY, Abecassis N, et al. Gastrointestinal stromal tumors: ESMO-EUROCAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2022; 33:20-33
Casali P, Le Cesne A, Poveda Velasco A, et al. Time to Definitive Failure to the First Tyrosine Kinase Inhibitor in Localized GI Stromal Tumors Treated With Imatinib As an Adjuvant: A European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Intergroup Randomized Trial in Collaboration With the Australasian Gastro-Intestinal Trials Group, UNICANCER, French Sarcoma Group, Italian Sarcoma Group, and Spanish Group for Research on Sarcomas. J Clin Oncol, 2015, 20 : 4276-83.
Casali P, Zalcberg J, Le Cesne A et al. Ten-year Progression-Free and Overall Survival in Patients with Unresectable or Metastatic GIST: Long-terme Analysis of the EORTC, ISG and AGTG intergroup Phase III Randomized Trial on Imatinib at Two Doses Levels. J Clin Oncol 2017 ; 35(15) : 1713-1720
Cassier PA, Fumagalli E, Rutkowski P et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012; 18:4458-64
Chi P, Shen S, Li J et al. A first-in-human phase 1 trial of NB003, a potent and selective KIT/PDGFRα inhibitor in patients with advanced GIST. ESMO® 2023, Abst #1916MO
Choi H Critical Issues in Response Evaluation on Computed Tomography: Lessons from the GIST Model. Curr Oncol Rep 2005 ; 7(4) : 307-311
Choi H, Charnsangavej C, Faria SC et al. Correlation of CT and PET in Patients with Metastatic GIST treated at a single Institution with Imatinib Mesylate: Proposal of New CT response criteria. J Clin Oncol 2007 ; 25(13) : 1753-9.
Corless CL, Barnett CM and Heinrich MC. GIST: Origin and molecular oncology. Nat Rev Oncol 2011; 11(12) : 865-78
Corless CL, Ballman KV, Antonescu CR et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol 2014; 32:1563-70
Cousin S, Bellera C, Guegan J et al. Regommune: a phase II study of regorafenib plus avelumab in solid tumors – Results of the advanced or metastatic GIST cohort, ESMO® 2023 Abstr #1920MO
Cuvelier C, Brahmi M, SobhaniI et al, Clinical description and development of a prognosis score for neurofibromatosis type 1 (NF1) associated GISTs in the RECKGIST cohort: a retrospective study from the French NETSARC+ network. ESMO 2024 Poster 1761P doi : 10.1016/j.annonc.2024.08.1852
De Pas T, Casali PG, Toma S et al. GIST: should they be treated with the same systemic chemotherapy as other soft tissue sarcomas ? Oncology 2003; 64:186–8
Dematteo RP, Ballman KV, Antonescu CR et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:1097-104
Demetri GD, Reichardt P, Kang YK et al. Efficacity and safety of regorafenib for advanced gastrointestinal stromal tumors after failure of imatinib and sunitinib. An international, multicentre, randomized, placebo-controlled phase 3 trial. Lancet 2013; 381:295-302
Demetri GD, Von Mehren M, Blanke CD et al. Efficacy and safety of imatinib in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347:472-80.
Demetri GD, Wang Y, Wehrle E et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol 2009; 27:3141-7
Demetri GD, Van Oosterom AT, Garrett CR et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006; 368: 1329-38
Drilon A, Laetsch TW, Kummar S et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 2018 22;378:731-739
Du CY, Zhou Y, Song C, et al. Is there a role of surgery in patients with recurrent or metastatic gastrointestinal stromal tumours responding to imatinib: a prospective randomised trial in China.Eur J Cancer 2014;50:1772-8
Ducimetiere F, Lurkin A, Ranchère-Vince D et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS ONE 2011 ; 6(8) e20294
Duffaud F, Meeus P, Bachet JB et al. Conservative surgery vs. duodeneopancreatectomy in primary duodenal gastrointestinal stromal tumors (GIST): a retrospective review of 114 patients from the French sarcoma group (FSG). Eur J Surg Oncol. 2014 ; 40(10): 1369-75
Emile JF, Brahimi S, Coindre JM et al. Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Med Oncol 2012; 29:1765-72.
Eisenberg BL, Harris J, Blanke C et al. Phase II Trial of Neoadjuvant/Adjuvant Imatinib Mesylate (IM) for Advanced Primary and Metastatic/Recurrent Operable Gastrointestinal Stromal Tumor (GIST) – early results of RTOG 0132. J Surg Oncol. 2009 Jan 1; 99(1): 42–47
Eriksson M, Reichardt P, Sundby HK et al. Needle biopsy through the abdominal wall for the diagnosis of gastrointestinal stromal tumour - Does it increase the risk for tumour cell seeding and recurrence? Eur J Cancer 2016 ; 59:128-133
Fletcher CDM, Berman J, Corless C et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002; 33:459-65.
Gasparotto D, Rossi S, Polano M et al. Quadruple-Negative GIST is a sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin Cancer Res 2017 ; 23(1) :273-282
George S, Blay JY, Casali PG et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer 2009; 45:1959-68
Giger OT, Ten Hoopen R, Shorthouse D et al. Preferential MGMT hypermethylation in SDH-deficient wild-type GIST. J Clin Pathol 2023; 77(1) : 34-39
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol 2010;28: 1247-53
Gronchi A, Bonvalot S, Poveda Velasco A et al. Quality of Surgery and Outcome in Localized Gastrointestinal Stromal Tumors Treated Within an International Intergroup Randomized Clinical Trial of Adjuvant Imatinib. JAMA Surg. 2020;155: e200397
Hautefeuille V, Cuvelier C, Sobhani I, et al. Impact of adjuvant imatinib on recurrence for neurofibromatosis type 1 (NF1) associated GISTs: an analysis of the RECKGIST cohort from the French NETSARC+ network.ESMO 2024 Poster 1760P doi : 10.1016/j.annonc.2024.08.1851
Heinrich MC, Jones RL, George S et al. Ripretinib versus sunitinib in gastrointestinal stromal tumor: ctDNA biomarker analysis of the phase 3 INTRIGUE trial Nature Medecine e-pub Jan 2024
Heinrich MC, Maki RG, Corless CL et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008; 26:5352-9
Jianchang W, Junbin Z, Zhuanpeng C et al. Survival outcome of local vs. radical excision in rectal gastrointestinal stromal tumor: a SEER database analysis. BMC Surgery 2022; 22:21
Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008;39: 1411-1419
Joensuu H, Vehtari A, Riihimäki J et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol 2012; 13:265-74.
Joensuu H, Rutkowski P, Nishida T et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol 2015; 33:634-42
Joensuu H, Eriksson M, Sundby Hall K, et al. One versus Three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA 2012; 307:1265-1272
Joensuu H, Eriksson M, Sundby Hall K, et al. Radiotherapy for GIST progressing during or after tyrosine kinase inhibitor therapy: A prospective study. J Clin Oncol 2016; 34: 244-50
Joensuu H, Eriksson M, Sundby Hall K, et al Three versus one year of adjuvant imatinib for high-risk gastrointestinal stromal tumor (GIST): Survival analysis of a randomized trial after 10 years of follow-up. Adjuvant Imatinib for High-Risk GI Stromal Tumor: Analysis of a Randomized Trial. J Clin Oncol 2020;38: Abs11503
Jones RL, Serrana C, von Mehren M et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: long-term efficacy and safety data from the NAVIGATOR phase 1 trial Eur J Cancer 2021;145:132-142
Kang YK, Ryu MH, Ryoo BY et al. Resumption of imatinib to control metastatic or unresectable GIST after failure of imatinib and sunitinib (RIGHT) : a randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2013 ; 14(12) : 1175-82
Kwak HV, Tardy KJ, Allbee A et al. Surgical Management of Germline GIST. Ann Surg Oncol 2023; 30(8) : 4966-4974
Landi B, Bouché O, Guimbaud R, Chayvialle JA. Gastrointestinal stromal tumors (GIST) <5 cm in size: review of the literature and expert propositions for clinical management. Gastroenterol Clin Biol 2010;3 4:120-33
Lartigue L, Neuville A, Lagarde P et al. Genomic Index predicts clinical outcome of intemediate-risk GIST providing a new inclusion criteria for imatinib adjuvant therapy. European Journal of Cancer 51 (2015) 75–83
Le Cesne A, Cropet C, Brahmi M, et al. LENVAGIST : a multicentre, comparative, placebo (P)-controlled, double-blinded, phase II study of the efficacy of Lenvatinib (L) in patients with advanced GIST after failure of imatinib and sunitinib. ESMO 2024 LBA79 doi : 10.1016/j.annonc.2024.08.2322
Marchiò C, Scaltriti M, Ladanyi M et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann Oncol. 2019; 30:1417-1427
Miettinen M and Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23:70-83
Mir O, Coriat R, Paci A et al. Impact of Proton Pump Inhibitors on Sunitinib Pharmacokinetics and Activity in GIST patients. ASCO 2018 Abstr 11538 - J Clin Oncology 36(15_suppl): 11538-11538
Naito Y, Komatsu Y, Kurukawa Y et al. CHAPTER-GIST-10: a phase1 study of pimitespib combined with imatinib in patients with imatinib-refractory GIST. ESMO® 2023, Abst #1917MO
Nannini M, Urbini M, Astolfi A et al. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J Transl Med 2017; 15:113
Nishida T, Naito Y, Takahashi T et al. Molecular and Clinicopathological features of KIT/PDGFR wild-type gastrointestinal tumors. Cancer Science 2024 ; 115: 894-904
Nishida T, Hølmebakk T, Raut CP et al. Defining Tumor Rupture in Gastrointestinal Stromal Tumor. Ann Surg Oncol 2019; 26:1669-1675
Patrikidou A, Domont J, Chabaud S et al. Long-term outcome of molecular subgroups of GIST patients treated with standard-dose imatinib in the BRF14 trial of the French Sarcoma Group. Eur J Cancer 2016 ; 52 :173-80
Piessen G, Lefèvre JH, Cabau M et al. Laparoscopic Versus Open Surgery for Gastric Gastrointestinal Stromal Tumors: What Is the Impact on Postoperative Outcome and Oncologic Results? Ann Surg 2015; 262:831-9.
Raut CP, Espat NJ, Maki RG et al. Efficay and Tolerability of 5-year Adjuvant Imatinib Treatment forPAtients with Resected Intermediate- or High-Risk Primary GIST: the PERSIST-5 Clinical Trial. JAMA Oncol 2018 vol 4(12) e184060
Ricci R. Syndromic gastrointestinal stromal tumors. Hered Cancer Clin Pract. 2016;14:15. doi: 10.1186/s13053-016-0055-4.
Robb WB, Bruyere E, Amielh D et al. Esophageal gastrointestinal stromal tumor: is tumoral enucleation a viable therapeutic option? Ann Surg 2015; 261:117-24.
Roulleaux Dugag M, Jones RL, Trent J et al. Beyond the driver Mutation: Immunotherapies in GIST. Front Immunol 2021 ; 12 p. 715727
Rutkowski P, Gronchi A, Hohenberger P et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol 2013; 20: 2937–2943
Shu P, Sun XF, Fang Y et al. Clinical outcomes of different therapeutic modalities for rectal gastrointestinal stromal tumor: Summary of 14-year clinical experience in a single center International Journal of Surgery 2020; 77 : 1–7
Soreide K, Sandvik OM, Soreide JA et al. Global epidemiology of gastrointestinal stromal tumours (GIST): a systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40: 39–46
Trent JC, Serrano C et al. Multi-Omic characterization of GIST in a large real-world cohort ASCO 2023
Van der Kleij M, Guchelaar N, Mathijssen R et al. Therapeutic Drug Monitoring of Kinase Inhibitors in Oncology. Clinical Pharmacokinetics 2023 62: 1333–1364
Verweij J, Casali PG, J Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364: 1127-34
Vincenzi B, Napoliatano A, Fioccio M et al. Adjuvant Imatinib in Patients with GIST Harboring Exon 9 Mutations: Results from a Multi-institutional European Retrospective Study. Clin Cancer Res, 2022 vol 28(8) pp 1672-79
Von Mehren M, Randall RL, Benjamin RS, et al. Gastrointestinal stromal tumors, version 2.2014. J Natl Compr Canc Netw 2014; 12:853-6
Von Mehren M, Kane JM, Riedel RF et al. Gastrointestinal Stromal Tumors, Version 2.2022 Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw 2022; 20(11):1204–1214
Yegin EG and Duman DG Small EUS-suspected gastrointestinal stromal tumors of the stomach: An overview for the current state of management. Endosc Ultrasound 2016; 5(2): 69-77
Zalcberg JR, Heinrich MC, Georges S et al. Clinical Benefit of Ripretinib Dose Escalation After Disease Progression in Advanced Gastrointestinal Stromal Tumor: An Analysis of the INVICTUS Study. Oncologist 2021; 26(11) : e2053-2060