18. Tumeurs desmoïdes de localisation abdominale ou associées à une polypose adénomateuse
(Dernière mise à jour le : )Principaux changements de la mise à jour du 25/01/2025
18.3.3. et 18.9. recours au réseau national spécialisé Poldigena pour prise en charge des polyposes digestives peut aider en cas de doute (https://www.fimatho.fr/poldigena).
18.4.3.2. et 18.7. Thérapie « ciblée »
• Le Desmoid Working Group recommande en 2024 l’usage en 1er lieu des traitements les moins toxiques suivant une évaluation au cas par cas du rapport bénéfice/risque sans standard thérapeutique spécifique
• Suite aux résultats de l’étude de phase 3 DeFi, OPTION du nirogacestat en accès précoce en 2ème ligne pour les malades avec TD réfractaire ou récidivante (risque d’aménorrhée secondaire)
18.4.4.2. essais cliniques : ouverture de l’étude CRYODESMO-2 phase II cryothérapie versus traitement médical par chimio. à base de méthotrexate et de vinblastine ou vinorelbine orale pour tumeur desmoïde extrapéritonéale.
18.5.2. la radiothérapie n’est pas recommandée chez l’enfant.
Groupe de travail et relecteurs
Groupe de travail :
Nicolas Benech (Lyon), coordonnateur,
Sylvie Bonvalot (Paris), Armelle Dufresne (Lyon), Afshin Gangi (Strasbourg), Thierry Lecomte (Tours), Cécile Le Péchoux (Villejuif), Daniel Lopez-Trabada-Ataz (Paris), Alexandra Meurgey (Lyon), Nicolas Penel (Lille), Sébastien Salas (Marseille), Daniel Orbach (Paris), Nayla Nicolas (Paris), Jean-Christophe Saurin (Lyon), Thomas Walter (Lyon).
Relecteurs :
Thomas Aparicio (Paris), Olivier Bouché (Reims), Mathilde Brasseur (Reims), Sylvain Causeret (Dijon), Olivier Collard (Saint-Priest-en-Jarez), André Dabrowski (Blendecques), Thérèse Delebecq-Janecki (Roubaix), Aurélien Dupré (Lyon), Sébastien Gaujoux (Paris), Boris Guiu (Montpellier), Florence Huguet (Paris), Marine Jary (Besançon), Dine Koriche (Béthune, Lens), Christophe Louvet (Paris), Frédéric Marchal (Nancy), Pascale Mariani (Paris), Pierre Meeus (Lyon), Nicolas Mocellin (Thionville), Jean-François Pailtel (Fréjus), Cornel Popovici (Marseille), Marc Pracht (Rennes).
Comment citer ce chapitre du TNCD ?
Benech N, Bonvalot S, Dufresne A, Gangi A, Le Péchoux C, Lopez-Trabada-Ataz D, Meurgey A, Nicolas N, Orbach D, Penel N, Salas S, Saurin JC, Walter T, Lecomte T, Bouché O; Thésaurus National de Cancérologie Digestive (TNCD);. Desmoid tumors located in the abdomen or associated with adenomatous polyposis: French intergroup clinical practice guidelines for diagnosis, treatment, and follow-up (SNFGE, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, ACHBT, SFR). Dig Liver Dis. 2022 Jun;54(6):737-746. doi: 10.1016/j.dld.2022.03.004. Epub 2022 May 1 PMID: 35508462.
Et mise à jour 2025 :
Benech N, Bonvalot S, Dufresne A, Gangi A, Le Péchoux C, Lopez-Trabada-Ataz D, Meurgey A, Penel N, Salas S, Orbach D, Nicolas N, Saurin JC, Walter T, Lecomte T, Bouché O. « Tumeurs desmoïdes ». Thésaurus National de Cancérologie Digestive, janvier 2025, en ligne [http://www.tncd.org]
18.1. Méthodologie
Du fait de la rareté des tumeurs desmoïdes (TD) et de la variabilité des tableaux de présentation clinico-radiologiques, le niveau de preuve disponible pour documenter leur prise en charge est relativement faible. Toutefois la publication récente de travaux prospectifs de haut niveau de preuve a permis d’enrichir qualitativement la littérature sur ces tumeurs. Ce document a été rédigé sur la base d’avis d’experts et d’une revue de la littérature à partir de la base de données Medline interrogée avec les mots clés « desmoid tumor » ou « desmoid fibromatosis » au 01/01/2025.
Les présentes recommandations ont été gradées selon le niveau des preuves disponibles dans la littérature (grade A, B ou C), ou en cas de preuves insuffisantes selon l'accord ou avis d'experts.
18.2. Introduction

L’association « SOS DESMOIDE » fondée en 1998 poursuit depuis sa création trois objectifs majeurs : Rompre l’isolement des malades atteints de tumeur desmoïde, rassembler et partager les connaissances existantes (expériences vécues du patient et connaissances cliniques et scientifiques des médecins) et favoriser la recherche afin de mieux comprendre pour mieux soigner.
18.2.1. DEFINITION
La TD est définie au plan histologique par une prolifération monomorphe fibroblastique des parties molles, classée dans les tumeurs intermédiaires dans la classification OMS 2020, à malignité locale (tumeur infiltrante, invasive), sans potentiel métastatique (CDM et al., n.d.). Toutes les localisations sont possibles : paroi abdominale, membres et ceintures, tête et cou, sein, tumeurs pelviennes, rétro-péritonéales et mésentériques.
18.2.2. Donnees epidemiologiques
Au plan épidémiologique, il s’agit d’une pathologie rare avec environ 400 cas/an en France, principalement des femmes (2/3 des TDs), avec une incidence annuelle estimée à 5-6 cas/millions d’habitants d’après les données issues de NETSARC (réseau français de Centres référents de prise en charge des sarcomes et tumeurs des tissus mous). L’âge médian au diagnostic varie de 35 à 44 ans (extrêmes 6- 90 ans), avec moins de 2 % des cas diagnostiqués avant l’âge de 15 ans (Penel et al., 2016, Kasper et al., 2011).
Nous traiterons dans ce chapitre du diagnostic, du traitement et du suivi uniquement des :
- TDs de la paroi abdominale (environ 30 % des cas),
- TDs intra-abdominales et mésentériques (environ 20 %)
- TDs associée à la polypose adénomateuse familiale (PAF- 15 à 20 % des TDs).
18.2.3. CARACTERISTIQUES HISTOLOGIQUES
Les TDs sont constituées d’une prolifération de cellules fusiformes fibroblastiques et myofibroblastiques agencées en longs faisceaux dans un fond collagénique ondulé comportant des vaisseaux à parois fines parfois cerclés d’un œdème. Ces cellules comportent un noyau ovalaire à chromatine fine avec un ou deux petits nucléoles et un cytoplasme amphophile. Il n’y a pas d’atypie cytonucléaire. Les mitoses sont rares ou absentes. Il n’y a pas de nécrose. Cette prolifération infiltre le muscle strié adjacent et peut recruter sur le front d’invasion des éléments inflammatoires. Dans les localisations intra-abdominales ou pédiatriques, des formes chéloïdiennes, hypercellulaires, inflammatoires ou myxoïdes sont décrites.
D’un point de vue immunohistochimique, il existe une expression de l’Actine-muscle lisse. Le CD34, l’EMA, la PS100 et le MUC4 sont négatifs. Le marquage avec l’anticorps anti-β-caténine donne un marquage nucléaire témoin de la dérégulation de la voie Wnt/ β-Caténine. Il ne permet pas de sursoir à l’analyse moléculaire par manque de spécificité.
18.2.4. CARACTERISTIQUES MOLECULAIRES
Plus de 90 % des TDs sont de forme sporadique liées à des variants génétiques pathogènes somatiques activateurs au niveau de l’exon 3 du gène CTNNB1 (T41A, S45F, S45P, essentiellement), pathognomonique dans un contexte de tumeur fibro-mésenchymateuse.
L’absence de variant pathogène somatique du gène CTNNB1 notamment dans les formes intra-abdominales doit faire rechercher une forme syndromique liée à des mutations inactivatrices ou des délétions du gène suppresseur de tumeur APC.
Les variants pathogènes des gènes CTNNB1 et APC sont mutuellement exclusifs. Les tumeurs non mutées pour CTNNB1 et APC sont extrêmement rares et sont liées à d’autres mécanismes de dérégulation de la voie de signalisation (Trautmann et al., 2020, Crago et al., 2015, Colombo et al., 2018, Le Guellec et al., 2012). Ces formes « wild-type » sont à considérer avec prudence et les diagnostics différentiels anatomopathologiques doivent être exclus avec ré-évaluation en centre expert (Crago et al., 2015).
18.2.5. Formes cliniques et contextes de découverte habituels
Avec une taille médiane au diagnostic de 5,5 cm (extrêmes : 1-55 cm), le diagnostic de ces tumeurs se fait le plus souvent dans le cadre du bilan d’un syndrome de masse parfois douloureux et inflammatoire des parties molles ou de la paroi abdominale (grands droits notamment ; Penel et al., 2016). Pour les localisations de la paroi abdominale, l’association avec la grossesse ou le post-partum (jusqu’à 18 mois) est également fréquente avec des poussées douloureuses pouvant être rythmées par les cycles menstruels imposant d’éliminer une endométriose.
Les TDs se développent fréquemment dans les suites d’une intervention chirurgicale, notamment pour les patients avec une PAF, avec le développement de lésions multiples de la paroi abdominale et du mésentère dans les 18 mois qui suivent une colectomie prophylactique (Kasper et al., 2011, Church et al., 2008).
Dans de rares situations, le diagnostic est réalisé au décours d’une prise en charge chirurgicale dans le cadre d’une complication aiguë :
- Perforation/abcédation dans le tube digestif d’une masse mésentérique
- Occlusion digestive sur syndrome de masse
- Urétéro-hydronéphrose des masses pelviennes et mésentériques
L’évolution est imprévisible et caractérisée dans les 2 ans suivant le diagnostic par une régression spontanée (30- 50 %), une progression locale (~20-30 %) ou une stabilité en taille des lésions (~30 %) (Kasper et al., 2015).
18.3. Explorations diagnostiques et pré-thérapeutiques
(cf. 18.8.1 et 18.8.2 algorithmes 1 et 2)
18.3.1. BILAN DIAGNOSTIQUE
L’imagerie joue un rôle essentiel dans le diagnostic, le bilan d’extension local, la planification préopératoire et le suivi des TDs. L’évolutivité et l’extension locorégionale, potentiellement très agressives, sont les critères les plus importants à évaluer pour orienter le traitement (surveillance, chirurgie ou traitement médical) et nécessitent une ré-évaluation radiologique initialement rapprochée (Kasper et al., 2017). Ces tumeurs n’ont pas de potentiel métastatique et il n’est donc pas nécessaire de faire de bilan d’extension à distance (https://www.nccn.org/professionals/physician_gls). Les mensurations tumorales peuvent être effectuées en 1, 2 ou 3 dimensions même si RECIST 1.1 reste la référence pour les essais thérapeutiques. Le changement de densité/signal peut aussi être employé pour juger du degré de fibrose (Braschi-Amirfarzan et al., 2016).
18.3.1.1. Localisations abdominales pariétales
L’IRM évalue au mieux les limites et le degré d’infiltration de la tumeur au sein des muscles. La lésion se développe le long des fascias musculaires pouvant donner un aspect en « queue de comète ». L’extension le long des fascias est un élément important pour la planification chirurgicale. Le signal IRM des lésions est fonction de l’importance du contingent de fibrose collagène (en hyposignal T2 et T1 du faible de sa faible contenance en eau et se rehaussant faiblement et tardivement après injection de produit de contraste) comparativement aux contingents de fibrose jeune et de matrice extra-cellulaire myxoïde (en hypersignal T2, hypo/isosignal T1 et se rehaussant de façon homogène ; Lee et al., 2006). Une fois le diagnostic établi, l’injection de produit de contraste n’est pas nécessaire dans la surveillance radiologique.
A l’échographie, lorsqu’elle est réalisée, les TDs se présentent comme des masses tissulaires plus ou moins bien délimitées au sein d’un muscle ou le long d’une aponévrose, d’échostructure hétérogène avec des composantes hypo-échogènes et une atténuation postérieure témoignant d’une composante fibreuse intra-lésionnelle au sein d’un stroma iso- ou hyperéchogène.
Bien que non spécifique, l’identification de la composante fibreuse est un argument diagnostique fort en imagerie, a fortiori en cas de présentation clinique évocatrice, notamment après césarienne ou traumatisme local, ou en contexte génétique de PAF.
18.3.1.2. Localisations intra-abdominales (rétro-péritonéales et mésentériques)
La TDM est l’examen de choix pour le diagnostic des complications (occlusion et perforation intestinale, ischémie mésentérique). Les TDs intra-abdominales apparaissent comme des lésions tissulaires plus ou moins bien délimitées, de contours spiculés, de rehaussement hétérogène, sans contingent nécrotique ou calcification intra-lésionnelle (Shinagare et al., 2011). Dans le cadre de la PAF, l’apparition d’une lésion mésentérique ou rétro-péritonéale présentant ces caractéristiques est fortement évocatrice de TD (Sinha et al., 2012). Les éléments importants à définir en pré-opératoire sont la taille de la lésion, ses rapports avec les vaisseaux mésentériques (notamment les branches proximales, permettant d’évaluer le risque de grêle court postopératoire), ses rapports avec les vaisseaux rétro-péritonéaux, et le degré d’infiltration des viscères adjacentes (grêle notamment). L’étendue de l’infiltration mésentérique de la lésion est souvent sous-estimée par l’imagerie.
18.3.1.3. Localisations extra-abdominales associées aux polyposes digestives
Les TDs associées à la PAF peuvent être multifocales, intra et/ou extra-abdominales. Les localisations extra-abdominales se situent majoritairement au niveau de la paroi (muscles grands droits de l’abdomen, orifice de stomie). Chez les enfants et les adolescents et adultes jeunes, la PAF peut être révélée par la présence d’une TD para-vertébrale lombaire classiquement appelé « fibrome de Gardner ». Dans ces localisations para-vertébrales, l’infiltration de l’os adjacent (donnant un aspect de scalloping osseux), est un signe d’agressivité locale progressive. Le bilan diagnostique ainsi que le suivi radiologique des patients atteints de PAF se fait essentiellement par IRM (dans le cas de lésion intra-abdominale associée ou de tumeurs intestinales), ou par scanner dans le contexte de l’urgence. Les TD postopératoires surviennent principalement dans les 5 ans qui suivent la colectomie. Afin de détecter les TDs dont la localisation met en danger la vie du patient, une palpation abdominale annuelle systématique est recommandée. En cas d'antécédents personnels ou familiaux de TD, une IRM ou un scanner abdominal doit être réalisé dans les 1 à 3 ans après la colectomie, puis à intervalles de 5 à 10 ans ou en cas de symptômes abdominaux évocateurs (National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. Https://Www.Nccn.Org/Professionals/Physician_gls (Accessed on October 14, 2020)., n.d.).
REFERENCES (avis d’experts)
- L’IRM est la modalité d’imagerie de référence des tumeurs desmoïdes.
- Le scanner abdomino-pelvien est l’examen de choix en cas de complication chirurgicale. Il peut également être utilisé pour permettre une meilleure visualisation des rapports vasculaires notamment pour les localisations intra-abdominales.
- L’étendue de l’infiltration mésentérique de la lésion est souvent sous-estimée par l’imagerie.
OPTION
- Échographie des parties molles dans le cadre d’une patiente surveillée dans un contexte de grossesse ou pour les petites lésions pariétales.
18.3.2. QUAND ET COMMENT REALISER UNE BIOPSIE ?
Conformément aux référentiels de l’European Society of Medical Oncology et au consensus Européen, le standard est de procéder lorsque cela est possible (taille tumorale, localisation) à une biopsie percutanée avec des aiguilles co-axiales de taille suffisante (14 ou 16G), car le diagnostic ne peut se faire sur la cytologie mais nécessite un examen histologique (Casali et al., 2018, Kasper et al., 2015). Une analyse moléculaire à la recherche de variant pathogène du gène de la ß-caténine (CTNNB1) ou APC est à réaliser systématiquement (Crago et al., 2015). La relecture dans le réseau de relecture de pathologie des sarcomes est également recommandée de manière systématique (https://netsarc.sarcomabcb.org).
Chez un patient atteint de polypose et ayant déjà le plus souvent une colectomie subtotale ou une colo-proctectomie, l’apparition d’une masse mésentérique, éventuellement au niveau du méso du réservoir iléal, est très évocatrice de TD (Church et al., 2015). Dans cette situation, une biopsie ne doit être réalisée qu'en cas de forte suspicion d'un diagnostic différentiel tel qu'un lymphome ou un carcinome. En particulier, la biopsie chirurgicale (par coelioscopie si possible) doit être strictement limitée dans cette situation car elle peut augmenter le risque de progression de la tumeur. Si celle-ci est suffisamment volumineuse, en cas d’indication de biopsie, on privilégiera une biopsie par voie percutanée. La voie endoscopique au moyen d’une écho-endoscopie basse est discutée en fonction de la localisation lorsque la voie percutanée n’est pas possible. Elle présente l’inconvénient de ne rapporter que peu de matériel, ce qui limite les possibilités de diagnostic et de biologie moléculaire. Une exérèse d’emblée à visée diagnostique est déconseillée du fait de son caractère préjudiciable au plan fonctionnel chez ces patients ayant déjà eu une colo-proctectomie.
Dans les cas sporadiques, il s’agit d’une masse pariétale, rétro péritonéale ou mésentérique, révélée par des signes digestifs non spécifiques ou une complication mécanique. En dehors d’un contexte d’urgence, doit être discutée en Réunion de Concertation Pluridisciplinaire (RCP), la réalisation d’une biopsie per cutanée, qui est le standard lorsqu’elle est possible anatomiquement (taille tumorale, localisation). Si la biopsie percutanée ou par voie endoscopique n’est pas possible, que la lésion est résécable d’emblée sans préjudice digestif étendu, et qu’un lymphome a été écarté, on s’oriente vers une chirurgie d’exérèse en évitant toute effraction tumorale qui serait préjudiciable en cas de sarcome. Si la tumeur est non résécable et la biopsie per-cutanée non réalisable, on s’oriente vers une biopsie chirurgicale, si possible sous cœlioscopie, afin de déterminer l’histologie et d’orienter un traitement médical éventuel. L’examen extemporané est source d’erreur car il ne permet pas d’analyse immunohistochimique ou de biologie moléculaire. Il faut donc éviter de prendre toute décision potentiellement mutilante sur la base de cet examen (Kasper et al., 2015).
REFERENCES (avis d’experts)
- Une micro-biopsie percutanée (pas de cytologie) doit être réalisée lorsque cela est possible (aiguille de 14 ou 16G).
- Une analyse moléculaire à la recherche de variant pathogène somatique du gène CTNNB1 doit être réalisée systématiquement en première intention.
- Si cette recherche est négative, il faut rechercher un variant pathogène constitutionnel du gène APC.
- Une tumeur desmoïde sans mutation APC/CTNNB1 doit faire évoquer un diagnostic différentiel tumoral.
- Une relecture dans le réseau de relecture en pathologie des sarcomes est systématiquement recommandée (https://netsarc.sarcomabcb.org).
- La biopsie chirurgicale est susceptible d’entraîner une poussée évolutive, et doit être limitée aux situations où le diagnostic différentiel avec une éventuelle adénopathie ou un carcinome ne peut être précisé.
OPTION
- Dans un contexte de PAF liée au gène APC avérée, et une imagerie fortement évocatrice réalisée en centre expert, il est possible de ne pas réaliser de biopsie de la tumeur initiale, notamment si celle-ci est potentiellement morbide.
18.3.3. QUAND ET COMMENT évoquer une cause génétique ou une polypose familiale ?
Dans le cadre des polyposes adénomateuses, seuls les patients porteurs d’un variant pathogène du gène APC sont susceptibles de développer des TDs. Les polyposes liées au gène MUTYH ne sont pas concernées par cette manifestation phénotypique. Chez les patients présentant une mutation constitutionnelle du gène APC, le risque cumulé de développement d’une TD est d’environ 10 %, avec un sexe ratio proche de 1 (Gurbuz et al., 1994). Les facteurs favorisants sont très clairement un antécédent de chirurgie abdominale (65-85 % des séries), et un spectre de variants pathogènes du gène APC en aval du codon 1440 de l’exon 15 (formes familiales de TD, corrélation génotype-phénotype ; Caspari et al., 1995). Deux études rétrospectives observent un lien statistique entre le développement de TD et une chirurgie plus lourde du type colo-proctectomie avec anastomose iléo-anale par rapport à la réalisation d’une colectomie subtotale avec anastomose iléo-rectale (Walter et al., 2017, Saito et al., 2016).
Dans la situation inverse de découverte d’une TD sans histoire familiale ou personnelle de polypose adénomateuse colorectale, les données sont beaucoup moins étayées. La plus importante étude rétrospective d’origine danoise publiée en 2019 a étudiée 626 patients présentant une TD, avec un diagnostic de polypose chez 26 patients (4,1 % ; van Houdt et al., 2019). Dans cette étude, un sous-groupe de 161 patients avait réalisé une coloscopie en l’absence d’antécédent personnel ou familial de polypose retrouvant le même chiffre de 4 % de polypose (6 patients) parmi les patients avec une TD et ayant réalisé une exploration endoscopique. La découverte d’une polypose est associée statistiquement à l’âge (< 40 ans), à la localisation abdominale ou rétro-péritonéale des TDs, au caractère multiple des tumeurs et à une histoire familiale de polypose. Deux études plus anciennes publiées en 1986 et en 2016 rapportent une fréquence de 1,3 % (1/75, rectoscopie ou examen radiologique du côlon) à 4,7 % (3/63, rectoscopie et test génétique) de polyposes sur des examens systématiques (Reitamo et al., 1986, Koskenvuo et al., 2016). L’étude de 2016 retrouvait également une fréquence plus élevée de cas jeunes (< 40 ans), de tumeurs abdominales et de tumeurs multiples dans le groupe polypose familiale, avec un sex-ratio proche de 1 au contraire des tumeurs sporadiques.
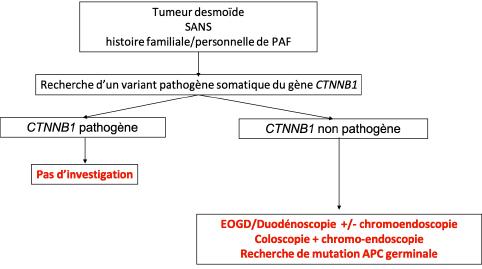
La biologie moléculaire des tumeurs peut être une aide à l’orientation diagnostique car une mutation de CTNBB1/ß-caténine semble exclure dans l’étude de 2016 la présence d’une polypose adénomateuse associée (82 cas, 0 polypose). Ce caractère exclusif de mutation de CTNNB1 sans polypose adénomateuse associée est rapporté dans une autre série, pédiatrique, de 44 TDs (29 cas mutés CTNNB1, 0 polypose). On peut donc proposer un algorithme décisionnel (ALGORITHME 1) pour la recherche d’une polypose familiale adénomateuse dans le cadre de la prise en charge initiale d’une TD (Wang et al., 2012). Le recours au réseau national spécialisé Poldigena pour la prise en charge des polyposes digestives de l’adulte peut aider en cas de doute (https://www.fimatho.fr/poldigena).
REFERENCES (niveau de recommandation grade C)
Il est recommandé de rechercher une polypose adénomateuse familiale associée à une tumeur desmoïde dans les cas suivants :
- ATCD familial de polypose adénomateuse
- Sexe masculin
- Age < 40 ans
- Présence de signes cliniques évocateurs d’un syndrome de Gardner : ostéome, kystes épidermiques cutanées, anomalie sur le fond d’œil…
- TD de localisations multiples
- Absence de variant pathogène somatique de CTNNB1 avec confirmation histologique du diagnostic
- Association à un variant pathogène somatique du gène APC.
- Le recours au réseau national spécialisé Poldigena pour la prise en charge des polyposes digestives de l’adulte peut aider en cas de doute (https://www.fimatho.fr/poldigena).
18.4. Traitements disponibles
18.4.1. Rationnel de la surveillance active en « première ligne »
La surveillance active des TDs a d’abord été proposée à des patients qui présentaient des récidives non résécables de manière conservatrice des membres (Lewis et al., 1999). En effet, s’agissant d’une tumeur bénigne, il n’y avait pas d’urgence à faire une amputation si les données de l’imagerie ne permettaient pas de proposer un traitement conservateur. L’évolution de ces patients a montré que la maladie se stabilisait dans plus de la moitié des cas, permettant alors d’éviter un geste mutilant. La surveillance initiale a ensuite été proposée à des patients présentant une tumeur primitive résécable (Bonvalot et al., 2008)(Fiore et al., 2009)(Colombo et al., 2015) (Duazo-Cassin et al., 2019) et il a été également montré que plus de la moitié des patients se stabilisaient ou régressaient spontanément. Ces données ont également été confirmées dans un travail colligeant 3 cohorte prospectives de TD extra-abdominales (Colombo et al., 2024). Passés trois ans, le risque d’évolutivité, en dehors d’un paramètre extérieur additionnel comme la grossesse, est très faible (Fiore et al., 2009). Cette stratégie a été évaluée dans différentes localisations, y compris intra-abdominales (Burtenshaw et al., 2016), puis dans des études rétrospectives de plus en plus larges, bénéficiant de plus de recul, avec une confirmation de ces résultats (Penel et al., 2016). De manière similaire, l’étude randomisée en double aveugle qui a comparé le sorafénib à un placebo chez des patients sélectionnés en raison d’une TD évolutive (récidive ou évolutivité confirmée par des examens successifs) a montré que dans le groupe placebo, les deux tiers des patients se stabilisaient ou présentaient une régression, dont 20 % étaient significatives selon RECIST1.1 (diminution d'au moins 30 % de la somme des diamètres des lésions cibles ; (M. M. Gounder et al., 2018). Les guidelines de l’ESMO et du consensus Européen recommandent désormais de commencer par une surveillance active ou un traitement médical seul des TDs avancées, en fonction de la symptomatologie (Casali et al., 2018), (Kasper et al., 2024).
18.4.2. Chirurgie, indications, modalités
18.4.2.1. Indications et modalités de la chirurgie d’urgence
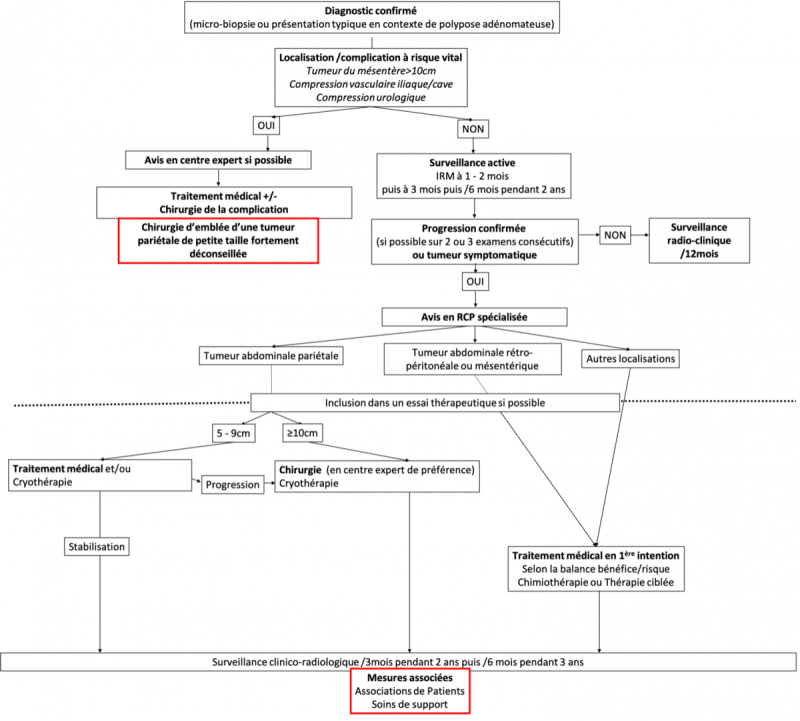
Les localisations intra-abdominales et en particulier mésentériques sont majoritairement celles (avec les localisations cervicales et médiastinales) qui sont responsables du décès éventuel des patients lors de la survenue de complications car une solution chirurgicale satisfaisante n’est pas toujours possible. Les patients qui présentent une complication chirurgicale révélatrice doivent bien évidemment être opérés. Il peut s’agir d’occlusion, de perforation digestive ou d’ischémie mésentérique. Si la résection de la tumeur n’est pas mutilante et n’impose qu’une résection digestive limitée, le traitement de la complication est fait en même temps que l’exérèse de la tumeur. Si l’exérèse de la tumeur entrainerait des sacrifices digestifs majeurs, il est préférable de traiter uniquement la complication et de laisser la tumeur en place, en la biopsiant pour avoir la confirmation histologique. Un traitement médical sera discuté en post opératoire. Les patients qui présentent un abcès pelvien ou des fistulisations survenant dans le contexte de polyposes déjà opérées seront traités préférentiellement par des drainages percutanés.
En l’absence de complication mécanique, une taille initiale importante au moment du diagnostic ou un risque vital en cas de progression ne sont pas en soi des indications opératoires immédiates après le diagnostic apporté par la biopsie, car on peut observer des régressions spontanées quelle que soit la taille de départ et la localisation. Dans les localisations ou la progression pourrait mettre en jeu la vie du patient, en particulier dans les localisations mésentériques, ou s’il s’y associent des symptômes, un traitement médical premier doit être discuté en RCP en centre expert (Casali et al., 2018, Kasper et al., 2015, Gronchi and Jones, 2019). La chirurgie n’a finalement que peu d’indications dans ces localisations défavorables avec un gros volume tumoral, car à la fois mutilante et exposée à un risque élevé de récidive et de complications. Par ailleurs, l’imagerie sous-évalue l’extension dans le mésentère car la lésion est mal limitée, si bien que la chirurgie est souvent macroscopiquement incomplète dans ces localisations. Dans tous les cas, ces décisions doivent être prises dans une RCP spécialisée.
Les récidives ont autant de chances de régresser que les tumeurs primitives et ne constituent pas non plus des indications de chirurgie immédiate (Fiore et al., 2009, Gounder et al., 2018). La chirurgie a en effet un rôle ambigu sur des reliquats macro ou microscopiques (qui ne sont pas différentes de chéloïdes sur le plan microscopique), car les facteurs de cicatrisation qu’elle entraine sont également des facteurs de croissance tumorale. Au fur et à mesure que l’on s’écarte d’une chirurgie suivie d’une rechute précoce, dans près de 50 % des cas la lésion se stabilise, voire régresse spontanément après une croissance initialement rapide.
18.4.2.2. Indications et modalités de la chirurgie d’urgence
La progression ou l’apparition de symptômes malgré un traitement médical adapté sont les principales indications opératoires potentielles. La première étape en cas de progression est de vérifier qu’il s’agit bien d’une TD, en particulier dans les localisations rétro-péritonéales. L’analyse moléculaire doit avoir été faite avec recherche de la mutation CTNNB1 du gène de la ß-caténine ou d’APC (Kasper et al., 2015). Avec les techniques de séquençage actuelles, les TDs non mutées sont finalement très rares (Crago et al., 2015). Les cas de diagnostic de « TD progressive » infirmé après relecture histologique en centre expert et analyse de biologie moléculaire sont fréquents. Cette relecture doit donc être systématique notamment en cas de traitement médical envisagé (Penel et al., 2016).
18.4.2.3. Quand discuter la chirurgie ?
L’indication d’un traitement local dépendra de la taille initiale, de la cinétique de progression, de la localisation, des symptômes et de l’âge du patient. Il est évident que pour une lésion initiale de petite taille (2 à 3 cm) et dans une localisation favorable (paroi abdominale par exemple), on pourra supporter un doublement de la taille de la tumeur sachant que l’on peut être au tout début de la phase de progression avec secondairement un ralentissement de celle-ci, puis une stabilisation voire une régression. La même taille initiale dans une localisation défavorable, à proximité d’axes vasculo-nerveux majeurs, incitera à un traitement plus rapidement. La courbe d’évolution est un élément central de la décision. Les patients doivent donc être surveillés de manière rapprochée après le diagnostic, car c’est à ce moment qu’il faut les catégoriser, puis on espace la surveillance si la tumeur est indolente ou se stabilise. On recommande un premier contrôle à un ou deux mois après l’imagerie initiale, puis trois mois plus tard si la maladie est stable puis six mois plus tard (ALGORITHME 2). Un traitement médical est discuté avant les traitements locaux invasifs, car ils peuvent stabiliser la tumeur et éviter des séquelles (Kasper et al., 2015). La décision d’opérer doit finalement être prise en RCP, et ne doit pas non plus être trop tardive en tenant compte de la courbe d’évolution de la TD.
18.4.2.4. Quelles sont les meilleures indications de la chirurgie en cas de progression ?
Les TDs de la paroi abdominale représentent une très bonne indication si la tumeur progresse de manière significative malgré un traitement médical (Bonvalot et al., 2008) en particulier lorsque la tumeur est ≥ 10 cm. Si la taille de la lésion se situe entre 5 et 10 cm et que celle-ci reste progressive entre deux imageries, on peut discuter soit la cryothérapie soit un traitement médical. En cas de poursuite de la progression, on s’oriente vers une chirurgie à type de pariétectomie. En effet, même si on considère qu’une pariétectomie abdominale est mutilante en particulier chez une femme jeune susceptible d’être enceinte, raison pour laquelle il est licite de bien sélectionner les patientes, une chirurgie complète est facilement accessible et reste fonctionnelle et bien supportée si la reconstruction est de qualité, et les grossesses à venir sont possibles même avec une plaque abdominale. Globalement, entre 15 et 20 % des patientes présentant une TD de la paroi abdominale finissent par être opérées (Bonvalot et al., 2013). La tumeur infiltrant le plus souvent le muscle dans toute son épaisseur, il s’agit en général d’une pariétectomie emportant le muscle atteint avec remplacement de la paroi par une plaque fixée en arrière des muscles restants. Sur le plan microscopique, l’impact des marges n’a été étudié que sur des séries anciennes qui mélangeaient des TDs indolentes et évolutives. Par ailleurs, le caractère mal limité de ces tumeurs et le nombre limité de coupes ne permet pas toujours d’être précis de manière rétrospective sur le caractère complet de la résection. On comprend qu’à mélanger des formes évolutives différentes avec des analyses anatomopathologiques plus ou moins précises, les études divergent quant à l’impact de ces marges. La chirurgie réglée s’adressant désormais à des tumeurs évolutives, il est possible que le fait d’avoir des marges négatives soit favorable, celles-ci étant pronostiques dans une étude multicentrique (Janssen et al., 2017).
Concernant les localisations rétro-péritonéales latérales, qui concernent fréquemment les vaisseaux iliaques et l’uretère, il ne faut envisager la chirurgie que lorsque celle-ci peut être macroscopiquement complète et en deuxième intention après un traitement médical d’induction. La radiothérapie est une alternative à discuter lorsqu’elle est possible et en tenant compte de l’âge du patient (cf. infra ; (Keus et al., 2013).
Concernant les localisations mésentériques, responsables d’ischémie et d’occlusion, la chirurgie est discutée si le traitement médical est inefficace, en privilégiant les situations où une exérèse complète peut être effectuée sans préjudice fonctionnel majeur. Des résections segmentaires digestives laissant en place des reliquats macroscopiques ne doivent être effectuées qu’au cas par cas après discussion en centre spécialisé.
REFERENCES (niveau de recommandation de grade A)
- La chirurgie n’est plus indiquée en 1ère ligne en dehors d’une complication mécanique.
- En cas de complication et de tumeur difficilement résécable ou avec un risque de chirurgie mutilante, la tumeur doit être laissée en place et le traitement chirurgical limité à la complication.
- La chirurgie hors urgence doit s’envisager en cas de localisation favorable (essentiellement pariétale). En dehors de ces localisations, l’indication doit être discutée en centre expert.
ESSAI CLINIQUE
Aucun
18.4.3. Traitements systémiques
Pendant de très nombreuses années ont été utilisés de multiples traitements systémiques, associés à des niveaux de preuve faibles issus de description de cas, de séries rétrospectives et d’études de phase II sans randomisation. Ces traitements sont simplement listés ici :
- anti-inflammatoires non stéroïdiens dont le sulindac, le celecoxib et le meloxicam (Nishida et al., 2010, Benech et al., 2017)
- tamoxifène, agonistes de la LHRH, anti-aromatase
- imatinib
- chimiothérapie : doxorubicine, doxorubicine pégylée, association doxorubicine-dacarbazine.(Penel et al., 2017, Kasper, 2015).
Deux essais cliniques randomisés ont été récemment publiés et permettent une meilleure évaluation du rapport bénéfice/risque à la fois de la chimiothérapie métronomique par l’association méthotrexate-vinblastine et les inhibiteurs de tyrosine kinase à large spectre (sorafénib et pazopanib).
18.4.3.1. Chimiothérapie cytotoxique
Les protocoles à base de Vinca-alcaloïde sont largement utilisés pour le traitement des TDs en particulier en administration intraveineuse. Dans un essai de phase II (n=30) non contrôlé, l’association méthotrexate-vinblastine administré une fois par semaine pendant 12-18 mois montrait un taux de réponse objective de 40% et de stabilité de 60 %. Après un suivi médian de 75 mois, la survie sans progression à 10 ans était de 67 % (Azzarelli et al., 2001). La tolérance et la toxicité à long terme étaient acceptables. Un essai de phase II (n=38) sur l’association méthotrexate (30 mg/m²) - vinblastine (6 mg/m²) toutes les 2 semaines chez des patients présentant une TD en progression a montré une survie sans progression à 5 ans de 80 %. Le taux de réponse objective était de 51%; de réponse partielle : 43 % ; de maladie stable : 5 %. Le délai médian pour obtenir une réponse objective était de 10 mois. Ce traitement a atténué la douleur dans 76 % des cas et a amélioré l’état fonctionnel dans 33 % des cas. Seulement 4 patients ont interrompu le traitement pour toxicité (Nishida et al., 2020). Mir et coll. ont également rapporté une série rétrospective de 90 patients traités avec la vinorelbine orale seule une fois par semaine (60-90 mg/m² ou une dose fixe de 90 mg). Les patients étaient fortement prétraités, avec un nombre médian de traitements systémiques de 2 (0-5), et 78 % d’entre eux recevait des opioïdes pour la gestion de la douleur. La durée médiane du traitement était de 6 mois. Les taux de survie sans progression à 6 et 12 mois étaient respectivement de 89 % et de 78 %. Le taux de réponse partielle était de 29 %, celui de stabilité de 57 %, et celui de progression de la maladie était de 14 % (Mir et al., 2020). Les protocoles à base de vinca-alcaloïdes à faible dose peuvent donc être considérés comme un traitement efficace, bénéficiant d’une évaluation favorable de la toxicité au long cours, ce qui est fondamental pour ces tumeurs bénignes, traitées chez des sujets jeunes.
Bien que moins évaluée, une chimiothérapie à base d’anthracyclines est une alternative possible avec toutefois une toxicité non négligeable.
18.4.3.2. Thérapie « ciblée »
Deux essais randomisés ont évalué le bénéfice d’inhibiteurs de tyrosine kinase à large spectre. Le 1er essai est un essai randomisé de phase II, non comparatif, évaluant d’une part le pazopanib et d’autre part l’association méthotrexate-vinblastine (Tableau 1 ; Toulmonde et al., 2019). Cet essai montre une certaine activité du pazopanib qui mériterait d’être évaluée formellement dans un essai comparatif. Le 2ème essai est un essai de phase 3 de supériorité comparant l’efficacité du sorafénib à la dose de 400 mg/jour contre un placebo. 87 patients ont été inclus. Il existait un avantage significatif en faveur du sorafénib. Cependant il convient de souligner que chez ces patients présentant des tumeurs évolutives ou menaçantes ou douloureuses, le placebo était associé à une réduction tumorale dans 20 % des cas (contre 33 % pour le sorafénib). Cet essai pourrait changer les pratiques, mais, il n’y a pas d’information sur la toxicité au long cours. La tolérance rapportée était également modérée avec 20 % des patients interrompant le traitement pour toxicité et un taux de toxicité de grade 3 de 20 %. Actuellement, aucune molécule ne dispose d’AMM dans cette indication.
En 2022, le nirogacestat, un inhibiteur oral de la γ-Secretase a été évalué dans un essai international multi-centrique de phase 3 contrôlé randomisé en double aveugle chez l’adulte avec TD progressive (étude DeFi, Tableau 1, (M. Gounder et al., 2023). Cent quarante-deux patients ont été inclus (70 traités par nirogacestat, 72 dans le bras placebo). Le pourcentage de patients ayant une réponse objective confirmée était de 41 % dans le groupe nirogacestat et de 8 % dans le groupe placebo (P<0,001). Des réponses complètes ont été observées chez 7 % des patients du groupe nirogacestat et chez aucun patient du groupe placebo. Le nirogacestat a montré un bénéfice significatif en termes de survie sans progression par rapport au placebo (HR : 0,29 ; IC 95 %[0,15-0,55] ; P<0,001). Il y avait une majorité de TD extra-abdominales (n=107), mais l’analyse en sous-groupe retrouvait une efficacité significative en considérant uniquement les patients avec TD intra-abdominales (n=35, HR : 0,17 ; IC95% [0,04–0,76]). Par ailleurs, 24 patients inclus présentaient une PAF avec une tendance à l'efficacité également observée dans ce sous-groupe même si la puissance statistique était insuffisante pour atteindre un seuil significatif de manière formelle (HR : 0,20 IC95% [0,04–1,00]). A noter que l’efficacité du nirogacestat concernait surtout les patients en ≥2ème ligne avec une maladie récidivante ou réfractaire après un précédent traitement (chirurgie, chimiothérapie, ou inhibiteur de tyrosine kinase ; HR : 0,30 ; IC95% [0,14 ; 0,63]). Une efficacité significative n’était pas observée chez les patients naïfs de traitement (HR : 0,77 IC95% [0,18–3,23]) bien que la puissance statistique de l’essai soit possiblement insuffisante pour l’évaluation du nirogacestat dans ce sous-groupe (n=32 patients).
Les événements indésirables fréquents associés au nirogacestat comprenaient la diarrhée (chez 84 % des patients), les nausées (chez 54 %), la fatigue (chez 51 %), l’hypophosphatémie (chez 42 %) et l’éruption maculopapuleuse (chez 32 %) ; 95 % des événements indésirables étaient de grade 1 ou 2. La survenue d’une aménorrhée secondaire était fréquente dans le bras nirogacestat avec 75 % des femmes en âge de procréer traitées présentant un dysfonctionnement ovarien (27/36), lequel s’est résolu en grande majorité par la suite (78 %, 21/27 dont 10/14 patientes ayant poursuivi le traitement ; (Loggers et al., 2024).
Suite à la publication de ces résultats le nirogacestat est disponible en accès précoce en France (https://ansm.sante.fr/tableau-acces-derogatoire/nirogacestat#). Ce traitement fait également l'objet d'une évaluation en cours chez les enfants atteints de TD (numéro ClinicalTrials.gov : NCT04195399).
La réponse au traitement systémique doit être évaluée idéalement par IRM (M. M. Gounder et al., 2018), (Ingley et al., 2019). Le Desmoid Working Group dans les recommandations internationales de 2024 recommande l’usage en 1er lieu des traitements les moins toxiques suivant une évaluation au cas par cas du rapport bénéfice/risque sans standard thérapeutique spécifique à recommander au vue des données actuelles de la littérature (Kasper et al., 2024). La durée maximale de traitement n’est pas définie concernant les thérapies ciblées.
REFERENCES
- Il n’y a pas de standard thérapeutique établi pour les tumeurs desmoïdes évolutives, nécessitant un traitement systémique (accord d'experts).
- La décision de traitement doit être discutée au cours d’une RCP spécialisée en centre expert en privilégiant les traitements les moins toxiques (accord d'experts).
OPTIONS
- Le profil d’efficacité et tolérance (notamment au long cours) de l’association méthotrexate-vinblastine est bien établie (niveau de recommandation de grade C).
- L’utilisation d’un inhibiteur de tyrosine kinase à large spectre (pazopanib ou sorafénib) est une option, dont la tolérance peut être difficile et dont la toxicité à long terme est mal établie (niveau de recommandation de grade C).
- L’utilisation du nirogacestat en accès précoce en 2ème ligne pour les malades avec TD réfractaire ou récidivante est une option, avec un risque d’aménorrhée secondaire chez la femme en âge de procréer (niveau de recommandation de grade B).
- L’utilisation d’AINS peut s’envisager notamment à visée antalgique (niveau de recommandation de grade C).
ESSAI CLINIQUE
Aucun
Tableau 1
Synthèse des essais contrôlés randomisés évaluant des traitements médicamenteux dans les tumeurs desmoïdes
| Essai | Gounder et al, 2018 | Toulmonde et al, 2019 | Gounder et al, 2023 |
|---|---|---|---|
| nombre de patients | 84 | 72 | 142 |
| Population | Patients avec une tumeur en progression : -Augmentation de la taille de la tumeur (≥ 10 % en 6 semaines) - Tumeur non résecable | Patients avec une tumeur en progression ≥ 20 % selon RECIST 1.1) sur les 6 derniers mois | Patients avec tumeur en progression ≥ 20 % selon RECIST 1.1) sur les 12 derniers mois |
| Intervention | Sorafénib (400 mg/d) jusqu’à intolérance ou progression | Pazopanib (800 mg/d) jusqu’à, intolérance ou progression ou jusqu’à 12 mois. | Nirogacestat oral (150 mg/12h), en continue par cycle de 28 jours en continue jusqu’à intolérance ou progression ou fin de l’essai. |
| Comparateur | Matched placebo | Methotrexate/Vinblastine IV pour 52 semaines | Matched placebo |
| Design | Essai de Phase 3 de supériorité (2:1 ratio) avec possible cross-over à progression | Essai de phase 2 non comparatif (2:1 ratio) avec cross-over | Essai de Phase 3 de supériorité (1 :1 ratio). |
| Critère de jugement principal | Absence de progression (PFS- RECIST 1.1 évaluation non centralisée) | Absence de progression à 6 mois (PFS- RECIST 1.1, évaluation centralisée pour le bras pazopanib) | Absence de progression définie sur l'imagerie ou critères cliniques, ou décès. (PFS- RECIST 1.1 et critères cliniques avec évaluation en aveugle centralisée) |
| Hypothèse statistique | 1-sided α=2.5 % ; β=10 % ; PFS (placebo) = 6 mois ; PFS (sorafénib) =1 5 mois ; HR=0.4 | Bras pazopanib : 1-sided α=5 %, β=20 %, H1=80 %, H0=60 % | Bras nirogacestat : 1-sided α=2.5 %, β=10 %, HR=0.4 |
| Résultats sur le critère de jugement principal | PFS median non atteinte dans le bras sorafénib HR pour la progression ou le décès dans le bras sorafénib =0.13 ; 95%IC : 0.05-0.31; P<0.001) | 6-mois PFS :
| PFS median non atteinte dans le bras nirogacestat. HR pour la progression ou le décès dans le bras nirogacestat = 0.29 ; IC95 %, 0,15 à 0,55 ; P<0,001). |
| Conclusion | Supériorité du sorafénib vs placebo | Pazopanib paraît un traitement prometteur | Nirogacestat paraît un traitement efficace sur les TD intra-abdominales et extra-abdominales en particulier en 2ème ligne. |
18.4.4. TRAITEMENTS LOCAUX
18.4.4.1. Radiothérapie
La radiothérapie n’a que peu d’indications dans les TDs abdominales. Si son utilisation a été validée dans une étude de phase 2, celle-ci a inclus surtout des localisations périphériques et à peine plus de 10 % de TD abdominale (Keus et al., 2013). Elle peut être discutée occasionnellement en centre expert sur des localisations pariétales abdominales récidivantes malgré les traitements validés : la situation clinique typique étant une TD de la paroi abdominale ayant progressé malgré un traitement médical bien suivi et/ou cryothérapie (cf. infra), opérée en deuxième intention par pariétectomie, et récidivante. L’autre indication potentielle à discuter en centre expert concerne, plus rarement, les TDs rétro-péritonéales ou intra-péritonéales progressives malgré le traitement médical, non résécables et dont le volume et la localisation permettent d’envisager une irradiation, dont les modalités techniques seront à définir au cas par cas. Dans tous les cas, cette indication exceptionnelle doit être validée en centre expert, en présence d'un onco-radiothérapeute.
18.4.4.2. Cryothérapie
La cryothérapie est une technique de destruction thermique par le froid (- 40 °C) des tumeurs. Dans le cas des TDs, elle est réalisée sous contrôle d'imagerie (TDM ou IRM). Elle est proposée aux patients avec TDs accessibles à un traitement percutané habituellement résistantes au traitement médical (deux lignes de traitement) avec progression tumorale et/ou symptomatique. Une première étude prospective non randomisées (CRYODESMO-01) a évalué la cryothérapie dans le traitement des TDs progressives sous traitement médical (cou, membres, thorax et paroi abdominale). Dans ce travail, 86 % (36/42) des patients avait une non-progression de leur maladie à 12 mois et une amélioration significative de l'état fonctionnel et de la douleur après traitement (Kurtz et al., 2021). La place de la cryothérapie dans la stratégie thérapeutique est évaluée dans le cadre du PHRC (CRYODESMO-02) qui va comparer de manière prospective et randomisée le traitement médical et la cryothérapie, avec la possibilité de proposer la cryothérapie en première ligne de traitement.
REFERENCES (avis d’experts)
- La radiothérapie est un traitement à envisager dans des indications exceptionnelles.
- Le traitement par cryothérapie en centre expert est une option thérapeutique en cours d’évaluation dans les tumeurs accessibles à un traitement percutané.
ESSAI CLINIQUE
- Etude CRYODESMO-2 : phase II randomisée évaluant la cryothérapie versus traitement médical par chimiothérapie à base de méthotrexate et de vinblastine ou vinorelbine orale pour le traitement de tumeur desmoïde extrapéritonéale. Coordonnateur : Pr A. GANGI (Strasbourg)
18.4.5. Autres mesures associées
18.4.5.1. Soins de support et d’accompagnement
Compte-tenu des douleurs, du préjudice fonctionnel, de la rareté de la maladie, et de l’incertitude concernant son évolution, le retentissement de la maladie peut être extrêmement sévère, avec :
- douleur ;
- impotence fonctionnelle ;
- anxiété, dépression ;
- difficultés professionnelles et financières ;
- isolement et incompréhension.
Les patients doivent pouvoir bénéficier de soins de support adaptés (Timbergen et al., 2018, Husson et al., 2019, Ingley et al., 2020, Rigaux et al., 2015). Le recours à des associations de soutien peut aider et doit être proposé : https://www.sos-desmoide.asso.fr/page/392623-actualites
Il n’y a pas de lien direct entre la douleur et l’évolutivité tumorale. Certaines tumeurs ayant spontanément régressées peuvent être source de douleurs importantes, a contrario, certaines tumeurs évolutives peuvent être totalement indolores. La douleur peut nécessiter une prise en charge algologique spécialisée.
18.4.5.2. Grossesse et contraception
La présence d’une TD en place n’est pas une contre-indication absolue à une grossesse ; il conviendra d’avoir si possible un recul évolutif de 2 ans, avec une maladie stable. Il faut savoir qu’il existe un risque de progression tumorale dans 30 % des cas en post-partum. La grossesse est possible mais nécessite un suivi par des équipes interdisciplinaires spécialisées (Fiore et al., 2014). Les imageries doivent être alors rapprochées (tous les 2 à 3 mois). L’échographie est la méthode de référence, selon les principes de radioprotection. Il existe un doute sur l’innocuité de la contraception orale notamment à base d’œstrogène ; si possible il convient de recommander une autre méthode de contraception.
18.4.5.3. Risques associés aux chirurgies
Tout traumatisme direct ou à distance (incluant chirurgie) est associé à un risque de poussée évolutive. Tout projet d’intervention chirurgicale doit être mis en perspective avec ce risque.
Dans le contexte d'une colectomie prophylactique pour les patients atteints de PAF, la chirurgie laparoscopique devra être préférée lorsque cela est possible car elle a été associée à un risque plus faible de TD postopératoire (Vitellaro et al., 2014).
REFERENCES (avis d’experts)
- La mise en relation avec une association de patient doit être systématiquement proposée de même que les soins d’accompagnement (douleur…).
- Tout projet d’intervention chirurgicale doit être mis en perspective avec un risque de poussée évolutive.
- Toute chirurgie esthétique ou tatouage/piercing doit être déconseillée.
- La grossesse est possible et nécessite un suivi par des équipes multidisciplinaires entraînées.
- Il existe un doute sur l’innocuité de la contraception orale notamment à base d’œstrogène ; si possible il convient de recommander une autre méthode de contraception.
18.5. Indications thérapeutiques (algorithme 2)
18.5.1. Prise en charge chez l’adulte (cf. 18.8.2. Algorithme 2)
18.5.2. Spécificité de prise en charge chez l’enfant
REFERENCES (niveau de recommandation de grade B)
- Les localisations abdominales et mésentériques sont rares chez l’enfant.
- Les données pédiatriques, principalement rétrospectives montrent que la prise en charge des tumeurs desmoïdes pédiatriques peut suivre de manière similaire les recommandations actuelles de cette maladie lorsqu’elle survient à l’âge adulte (Orbach et al., 2017).
- La stratégie d’observation première doit être favorisée si la position de la tumeur et son volume permettent cette surveillance sans risque pour l’enfant (Duhil de Bénazé et al., 2020, Sparber-Sauer et al., 2021).
- Les traitements sont indiqués en cas de tumeurs menaçantes, situées en site dangereux ou clairement progressives après une période de surveillance. Les gestes opératoires doivent aussi être évités au maximum.
- Les biopsies initiales doivent être réalisées préférentiellement par voie percutanée.
- Du fait de son absence d’effet indésirable connu à long terme, l’association méthotrexate-vinblastine reste la 1ère ligne de traitement médicamenteux en pédiatrie (Skapek et al., 2007).
- La radiothérapie n’est pas recommandé chez l’enfant (Kasper et al., 2024).
OPTIONS
- Les inhibiteurs des tyrosines kinases n’ont pas été évalués en pédiatrie, mais les toxicités potentielles, connues (thyroïde, cardiaque) ou non encore connues, limitent leur utilisation. Ils ne sont discutés comme traitement de seconde ligne qu’en cas d’échec thérapeutique des autres traitements et chez les patients les plus âgés.
- Les données concernant l’efficacité de l’Hydréa© (hydroxyurée) restent à confirmer (Ferrari et al., 2019).
- Les traitements hormonaux et par AINS ont montré une efficacité très modeste en pédiatrie (< 8 % de taux de réponse).
18.6. Surveillance
18.6.1. Suivi initial et post-thérapeutique (cf. 18.8.2. Algorithme 2)
A ce jour, il n’existe pas de recommandation spécifique pour le suivi des TDs (sporadiques ou associées à la PAF). Dans la plupart de cas, les TDs sont incluses dans le chapitre des sarcomes comme dans les recommandations NCCN, une fréquence standard de surveillance est proposée : la première imagerie après le diagnostic doit être rapprochée entre 1 et 2 mois puis une imagerie tous les 3 à 6 mois pendant les deux premières années, puis annuelle après (https://www.nccn.org/professionals/physician_gls).
Le groupe européen d’étude des TDs conseille la surveillance par IRM afin de diminuer l’exposition aux rayons X, avec une meilleure étude du signal de la lésion et de son infiltration (Kasper et al., 2017). Une surveillance par IRM sans contraste peut être envisagée lorsque le diagnostic est établi avec certitude afin de minimiser les risques pour le patient lié à l’injection de produit de contraste. En revanche, une évaluation par TDM avec injection de produits de contraste peut être préférée à l’IRM dans les localisations mésentériques à haut risque, car il présente une meilleure résolution pour évaluer le contact avec les vaisseaux et l’étude de la survenue possible de complications (obstruction intestinale…).
Il n’y a pas de consensus sur la durée de la surveillance au-delà des 5 premières années de suivi. En cas de stabilisation depuis plus de cinq ans d’une TD de bas risque, un arrêt de la surveillance radiologique peut être envisagé et ce même si la tumeur est en place (Ghert et al., 2014).
Les modalités de la surveillance peuvent être adaptées à la localisation et aux modalités de prise en charge précédemment entreprises : traitement curatif (rémission complète après traitement/chirurgie/cryothérapie) ou traitement ayant permis seulement de contrôler la progression tumorale (stabilisation maintenue après l’arrêt du traitement). Dans le cadre d’une PAF, la surveillance est à réaliser en association avec le suivi endoscopique digestif.
REFERENCES (avis d’experts)
- L‘IRM est l’examen de 1ère intention pour le suivi des tumeurs desmoïdes
- La première imagerie de suivi après le diagnostic doit être rapprochée entre 1 et 2 mois puis espacée en cas de stabilité tumorale à tous les 3 à 6 mois pendant les deux premières années, puis annuellement.
- Surveillance post-thérapeutique : examen clinique + imagerie (TDM/IRM) tous les 3 mois pendant 2 ans puis tous les 6 mois pendant 3 ans. Après cinq ans : évaluation une fois par an avec examen clinique +/- imagerie selon la localisation et la prise en charge initiale
- La survenue d’une grossesse chez les patientes avec une tumeur desmoïde en place ou résiduelle après traitement, doit nécessiter une surveillance radiologique rapprochée (tous les 2 à 3 mois) par échographie ou IRM.
18.6.2. Cohorte de suivi française
Cohorte ALTITUDES (fermée aux inclusions, suivi en cours) : « Prospective observational cohort of incident cases of DT diagnosed in France (Biobanking, quality of life data, outcome) ». Cohorte dont l’objectif est de réaliser un suivi prospectif des cas incidents de TD avec description de cas associés à la PAF, du pourcentage de variant pathogène du gène CTNNB1, des traitements reçus, de leur impact et des potentiels facteurs de réponse thérapeutique. La qualité de vie des patients est aussi évaluée. Le bilan d’inclusion comprenait une prise de sang pour évaluer la sensibilité et spécificité de certaines techniques d’extraction de l’ADN circulant ainsi que l’intérêt d’une coloscopie systématique pour le dépistage de néoplasie digestive associée.
18.7. PROTOCOLES DE TRAITEMENT SYSTEMIQUE POUR LES TUMEURS DESMOIDES EVOLUTIVES
Méthotrexate-Vinblastine
Méthotrexate IV 30 mg/m² tous les 7 ou 15 jours pendant 12-18mois
Vinblastine IV 6 mg/m² tous les 7 ou 15 jours pendant 12-18 mois
Vinorelbine orale
60 à 90 mg per os 1 fois par semaine pendant 6 mois à 12 mois
Sorafénib
400 mg par jour per os – durée maximale de traitement non codifiée
Pazopanib
800 mg par jour per os – durée maximale de traitement non codifiée
Nirogacestat
150 mg par jour per os – durée maximale de traitement non codifiée
18.8. ARBRES DECISIONNELS (ALGORITHMES)
18.8.1. ALGORITHME 1 - Stratégie diagnostique à la recherche d’une polypose adénomateuse familiale dans le cadre de la découverte d’une tumeur desmoïde
18.8.2. ALGORITHME 2 - Stratégie de prise en charge des tumeurs desmoïdes abdominales et/ou associées à une polypose adénomateuse
18.9. CENTRES EXPERTS
- Réseau NETSARC+ et réseau de relecture en pathologie des sarcomes : https://netsarc.sarcomabcb.org
- Centres experts reconnus dans la prise en charge des polyposes intestinales
- Réseau national spécialisé Poldigena pour la prise en charge des polyposes digestives de l’adulte (https://www.fimatho.fr/poldigena).
18.10. BIBLIOGRAPHIE
- Azzarelli, A., Gronchi, A., Bertulli, R., Tesoro, J. D., Baratti, D., Pennacchioli, E., Dileo, P., Rasponi, A., Ferrari, A., Pilotti, S., & Casali, P. G. (2001). Low-dose chemotherapy with methotrexate and vinblastine for patients with advanced aggressive fibromatosis. Cancer, 92(5), 1259–1264. https://doi.org/10.1002/1097-0142(20010901)92:5<1259::aid-cncr1446>3.0.co;2-y
- Benech, N., Walter, T., & Saurin, J.-C. (2017). Desmoid Tumors and Celecoxib with Sorafenib. The New England Journal of Medicine, 376(26), 2595–2597. https://doi.org/10.1056/NEJMc1702562
- Bonvalot, S., Eldweny, H., Haddad, V., Rimareix, F., Missenard, G., Oberlin, O., Vanel, D., Terrier, P., Blay, J. Y., Le Cesne, A., & Le Péchoux, C. (2008). Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. European Journal of Surgical Oncology: The Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 34(4), 462–468. https://doi.org/10.1016/j.ejso.2007.06.006
- Bonvalot, S., Ternès, N., Fiore, M., Bitsakou, G., Colombo, C., Honoré, C., Marrari, A., Le Cesne, A., Perrone, F., Dunant, A., & Gronchi, A. (2013). Spontaneous regression of primary abdominal wall desmoid tumors: More common than previously thought. Annals of Surgical Oncology, 20(13), 4096–4102. https://doi.org/10.1245/s10434-013-3197-x
- Braschi-Amirfarzan, M., Keraliya, A. R., Krajewski, K. M., Tirumani, S. H., Shinagare, A. B., Hornick, J. L., Baldini, E. H., George, S., Ramaiya, N. H., & Jagannathan, J. P. (2016). Role of Imaging in Management of Desmoid-type Fibromatosis: A Primer for Radiologists. Radiographics: A Review Publication of the Radiological Society of North America, Inc, 36(3), 767–782. https://doi.org/10.1148/rg.2016150153
- Burtenshaw, S. M., Cannell, A. J., McAlister, E. D., Siddique, S., Kandel, R., Blackstein, M. E., Swallow, C. J., & Gladdy, R. A. (2016). Toward Observation as First-line Management in Abdominal Desmoid Tumors. Annals of Surgical Oncology, 23(7), 2212–2219. https://doi.org/10.1245/s10434-016-5159-6
- Casali, P. G., Abecassis, N., Aro, H. T., Bauer, S., Biagini, R., Bielack, S., Bonvalot, S., Boukovinas, I., Bovee, J. V. M. G., Brodowicz, T., Broto, J. M., Buonadonna, A., De Álava, E., Dei Tos, A. P., Del Muro, X. G., Dileo, P., Eriksson, M., Fedenko, A., Ferraresi, V., … ESMO Guidelines Committee and EURACAN. (2018). Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology: Official Journal of the European Society for Medical Oncology, 29(Suppl 4), iv268–iv269. https://doi.org/10.1093/annonc/mdy321
- Caspari, R., Olschwang, S., Friedl, W., Mandl, M., Boisson, C., Böker, T., Augustin, A., Kadmon, M., Möslein, G., & Thomas, G. (1995). Familial adenomatous polyposis: Desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Human Molecular Genetics, 4(3), 337–340. https://doi.org/10.1093/hmg/4.3.337
- CDM, F., JA, B., PCW, H., & F, M. (n.d.). WHO Classification of Tumours of Soft Tissue and Bone. Retrieved 10 March 2021, from https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/WHO-Classification-Of-Tumours-Of-Soft-Tissue-And-Bone-2013
- Church, J., Lynch, C., Neary, P., LaGuardia, L., & Elayi, E. (2008). A desmoid tumor-staging system separates patients with intra-abdominal, familial adenomatous polyposis-associated desmoid disease by behavior and prognosis. Diseases of the Colon and Rectum, 51(6), 897–901. https://doi.org/10.1007/s10350-008-9232-5
- Church, J., Xhaja, X., LaGuardia, L., O’Malley, M., Burke, C., & Kalady, M. (2015). Desmoids and genotype in familial adenomatous polyposis. Diseases of the Colon and Rectum, 58(4), 444–448. https://doi.org/10.1097/DCR.0000000000000316
- Colombo, C., Hakkesteegt, S., Le Cesne, A., Barretta, F., Blay, J.-Y., Grünhagen, D. J., Penel, N., Lam, L., Fiore, M., Palassini, E., Grignani, G., Tolomeo, F., Collini, P., Merlini, A., Perrone, F., Stacchiotti, S., Verhoef, C., Bonvalot, S., & Gronchi, A. (2024). Active surveillance in patients with extra-abdominal desmoid-type fibromatosis: A pooled analysis of three prospective observational studies. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. https://doi.org/10.1158/1078-0432.CCR-24-2340
- Colombo, C., Miceli, R., Le Péchoux, C., Palassini, E., Honoré, C., Stacchiotti, S., Mir, O., Casali, P. G., Dômont, J., Fiore, M., Le Cesne, A., Gronchi, A., & Bonvalot, S. (2015). Sporadic extra abdominal wall desmoid-type fibromatosis: Surgical resection can be safely limited to a minority of patients. European Journal of Cancer (Oxford, England: 1990), 51(2), 186–192. https://doi.org/10.1016/j.ejca.2014.11.019
- Colombo, C., Urbini, M., Astolfi, A., Collini, P., Indio, V., Belfiore, A., Paielli, N., Perrone, F., Tarantino, G., Palassini, E., Fiore, M., Pession, A., Stacchiotti, S., Pantaleo, M. A., & Gronchi, A. (2018). Novel intra-genic large deletions of CTNNB1 gene identified in WT desmoid-type fibromatosis. Genes, Chromosomes & Cancer, 57(10), 495–503. https://doi.org/10.1002/gcc.22644
- Crago, A. M., Chmielecki, J., Rosenberg, M., O’Connor, R., Byrne, C., Wilder, F. G., Thorn, K., Agius, P., Kuk, D., Socci, N. D., Qin, L.-X., Meyerson, M., Hameed, M., & Singer, S. (2015). Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes, Chromosomes & Cancer, 54(10), 606–615. https://doi.org/10.1002/gcc.22272
- Duazo-Cassin, L., Le Guellec, S., Lusque, A., Chantalat, E., Laé, M., Terrier, P., Coindre, J.-M., Boulet, B., Le Boulc’h, M., Gangloff, D., Meresse, T., Chaput, B., Al Ali, A., Rimareix, F., Bonvalot, S., & Vaysse, C. (2019). Breast desmoid tumor management in France: Toward a new strategy. Breast Cancer Research and Treatment, 176(2), 329–335. https://doi.org/10.1007/s10549-019-05245-5
- Duhil de Bénazé, G., Vigan, M., Corradini, N., Minard-Colin, V., Marie-Cardine, A., Verite, C., Defachelles, A. S., Thebaud, E., Castex, M. P., Sirvent, N., Bodet, D., Mansuy, L., Rome, A., Petit, A., Plantaz, D., Jourdain, A., Mary, P., Carton, M., & Orbach, D. (2020). Functional analysis of young patients with desmoid-type fibromatosis: Initial surveillance does not jeopardize long term quality of life. European Journal of Surgical Oncology: The Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 46(7), 1294–1300. https://doi.org/10.1016/j.ejso.2020.02.028
- Ferrari, A., Orbach, D., Affinita, M. C., Chiaravalli, S., Corradini, N., Meazza, C., Bisogno, G., & Casanova, M. (2019). Evidence of hydroxyurea activity in children with pretreated desmoid-type fibromatosis: A new option in the armamentarium of systemic therapies. Pediatric Blood & Cancer, 66(1), e27472. https://doi.org/10.1002/pbc.27472
- Fiore, M., Coppola, S., Cannell, A. J., Colombo, C., Bertagnolli, M. M., George, S., Le Cesne, A., Gladdy, R. A., Casali, P. G., Swallow, C. J., Gronchi, A., Bonvalot, S., & Raut, C. P. (2014). Desmoid-type fibromatosis and pregnancy: A multi-institutional analysis of recurrence and obstetric risk. Annals of Surgery, 259(5), 973–978. https://doi.org/10.1097/SLA.0000000000000224
- Fiore, M., Rimareix, F., Mariani, L., Domont, J., Collini, P., Le Péchoux, C., Casali, P. G., Le Cesne, A., Gronchi, A., & Bonvalot, S. (2009). Desmoid-type fibromatosis: A front-line conservative approach to select patients for surgical treatment. Annals of Surgical Oncology, 16(9), 2587–2593. https://doi.org/10.1245/s10434-009-0586-2
- Ghert, M., Yao, X., Corbett, T., Gupta, A. A., Kandel, R. A., Verma, S., & Werier, J. (2014). Treatment and follow-up strategies in desmoid tumours: A practice guideline. Current Oncology (Toronto, Ont.), 21(4), e642-649. https://doi.org/10.3747/co.21.2112
- Gounder, M. M., Mahoney, M. R., Van Tine, B. A., Ravi, V., Attia, S., Deshpande, H. A., Gupta, A. A., Milhem, M. M., Conry, R. M., Movva, S., Pishvaian, M. J., Riedel, R. F., Sabagh, T., Tap, W. D., Horvat, N., Basch, E., Schwartz, L. H., Maki, R. G., Agaram, N. P., … Schwartz, G. K. (2018). Sorafenib for Advanced and Refractory Desmoid Tumors. The New England Journal of Medicine, 379(25), 2417–2428. https://doi.org/10.1056/NEJMoa1805052
- Gounder, M., Ratan, R., Alcindor, T., Schöffski, P., van der Graaf, W. T., Wilky, B. A., Riedel, R. F., Lim, A., Smith, L. M., Moody, S., Attia, S., Chawla, S., D’Amato, G., Federman, N., Merriam, P., Van Tine, B. A., Vincenzi, B., Benson, C., Bui, N. Q., … Kasper, B. (2023). Nirogacestat, a γ-Secretase Inhibitor for Desmoid Tumors. The New England Journal of Medicine, 388(10), 898–912. https://doi.org/10.1056/NEJMoa2210140
- Gronchi, A., & Jones, R. L. (2019). Treatment of Desmoid Tumors in 2019. JAMA Oncology, 5(4), 567–568. https://doi.org/10.1001/jamaoncol.2018.6449
- Gurbuz, A. K., Giardiello, F. M., Petersen, G. M., Krush, A. J., Offerhaus, G. J., Booker, S. V., Kerr, M. C., & Hamilton, S. R. (1994). Desmoid tumours in familial adenomatous polyposis. Gut, 35(3), 377–381. https://doi.org/10.1136/gut.35.3.377
- Husson, O., Younger, E., Dunlop, A., Dean, L., Strauss, D. C., Benson, C., Hayes, A. J., Miah, A., van Houdt, W., Zaidi, S., Smith, M., Williams, J., Jones, R. L., & van der Graaf, W. T. A. (2019). Desmoid fibromatosis through the patients’ eyes: Time to change the focus and organisation of care? Supportive Care in Cancer: Official Journal of the Multinational Association of Supportive Care in Cancer, 27(3), 965–980. https://doi.org/10.1007/s00520-018-4386-8
- Ingley, K. M., Burtenshaw, S. M., Theobalds, N. C., White, L. M., Blackstein, M. E., Gladdy, R. A., Thipphavong, S., & Gupta, A. A. (2019). Clinical benefit of methotrexate plus vinorelbine chemotherapy for desmoid fibromatosis (DF) and correlation of treatment response with MRI. Cancer Medicine, 8(11), 5047–5057. https://doi.org/10.1002/cam4.2374
- Ingley, K. M., Klein, R., Theobalds, N., Burtenshaw, S., Abdul Razak, A. R., Chen, B., Xu, W., Gladdy, R., Li, M., & Gupta, A. A. (2020). High prevalence of persistent emotional distress in desmoid tumor. Psycho-Oncology, 29(2), 311–320. https://doi.org/10.1002/pon.5250
- Janssen, M. L., van Broekhoven, D. L. M., Cates, J. M. M., Bramer, W. M., Nuyttens, J. J., Gronchi, A., Salas, S., Bonvalot, S., Grünhagen, D. J., & Verhoef, C. (2017). Meta-analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid-type fibromatosis. The British Journal of Surgery, 104(4), 347–357. https://doi.org/10.1002/bjs.10477
- Kasper, B. (2015). Systemic treatment approaches for sporadic desmoid-type fibromatosis: Scarce evidence and recommendations. Oncology Research and Treatment, 38(5), 244–248. https://doi.org/10.1159/000381909
- Kasper, B., Baldini, E. H., Bonvalot, S., Callegaro, D., Cardona, K., Colombo, C., Corradini, N., Crago, A. M., Dei Tos, A. P., Dileo, P., Elnekave, E., Erinjeri, J. P., Navid, F., Farma, J. M., Ferrari, A., Fiore, M., Gladdy, R. A., Gounder, M., Haas, R. L., … Desmoid Tumor Working Group. (2024). Current Management of Desmoid Tumors: A Review. JAMA Oncology, 10(8), 1121–1128. https://doi.org/10.1001/jamaoncol.2024.1805
- Kasper, B., Baumgarten, C., Bonvalot, S., Haas, R., Haller, F., Hohenberger, P., Moreau, G., van der Graaf, W. T. A., Gronchi, A., & Desmoid Working Group. (2015). Management of sporadic desmoid-type fibromatosis: A European consensus approach based on patients’ and professionals’ expertise - a sarcoma patients EuroNet and European Organisation for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group initiative. European Journal of Cancer (Oxford, England: 1990), 51(2), 127–136. https://doi.org/10.1016/j.ejca.2014.11.005
- Kasper, B., Baumgarten, C., Garcia, J., Bonvalot, S., Haas, R., Haller, F., Hohenberger, P., Penel, N., Messiou, C., van der Graaf, W. T., Gronchi, A., & Desmoid Working Group. (2017). An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Annals of Oncology: Official Journal of the European Society for Medical Oncology, 28(10), 2399–2408. https://doi.org/10.1093/annonc/mdx323
- Kasper, B., Ströbel, P., & Hohenberger, P. (2011). Desmoid tumors: Clinical features and treatment options for advanced disease. The Oncologist, 16(5), 682–693. https://doi.org/10.1634/theoncologist.2010-0281
- Keus, R. B., Nout, R. A., Blay, J.-Y., de Jong, J. M., Hennig, I., Saran, F., Hartmann, J. T., Sunyach, M. P., Gwyther, S. J., Ouali, M., Kirkpatrick, A., Poortmans, P. M., Hogendoorn, P. C. W., & van der Graaf, W. T. A. (2013). Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid-type fibromatosis—An EORTC STBSG and ROG study (EORTC 62991-22998). Annals of Oncology: Official Journal of the European Society for Medical Oncology, 24(10), 2672–2676. https://doi.org/10.1093/annonc/mdt254
- Koskenvuo, L., Peltomäki, P., Renkonen-Sinisalo, L., Gylling, A., Nieminen, T. T., Ristimäki, A., & Lepistö, A. (2016). Desmoid tumor patients carry an elevated risk of familial adenomatous polyposis. Journal of Surgical Oncology, 113(2), 209–212. https://doi.org/10.1002/jso.24117
- Kurtz, J.-E., Buy, X., Deschamps, F., Sauleau, E., Bouhamama, A., Toulmonde, M., Honoré, C., Bertucci, F., Brahmi, M., Chevreau, C., Duffaud, F., Gantzer, J., Garnon, J., Blay, J.-Y., & Gangi, A. (2021). CRYODESMO-O1: A prospective, open phase II study of cryoablation in desmoid tumour patients progressing after medical treatment. European Journal of Cancer (Oxford, England: 1990), 143, 78–87. https://doi.org/10.1016/j.ejca.2020.10.035
- Le Guellec, S., Soubeyran, I., Rochaix, P., Filleron, T., Neuville, A., Hostein, I., & Coindre, J.-M. (2012). CTNNB1 mutation analysis is a useful tool for the diagnosis of desmoid tumors: A study of 260 desmoid tumors and 191 potential morphologic mimics. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc, 25(12), 1551–1558. https://doi.org/10.1038/modpathol.2012.115
- Lee, J. C., Thomas, J. M., Phillips, S., Fisher, C., & Moskovic, E. (2006). Aggressive fibromatosis: MRI features with pathologic correlation. AJR. American Journal of Roentgenology, 186(1), 247–254. https://doi.org/10.2214/AJR.04.1674
- Lewis, J. J., Boland, P. J., Leung, D. H., Woodruff, J. M., & Brennan, M. F. (1999). The enigma of desmoid tumors. Annals of Surgery, 229(6), 866–872; discussion 872-873. https://doi.org/10.1097/00000658-199906000-00014
- Loggers, E. T., Chugh, R., Federman, N., Hartner, L., Riedel, R. F., Cho, S., Hyslop, D., Lim, A., Oton, A. B., & Oktay, K. H. (2024). Onset and resolution of ovarian toxicity with nirogacestat treatment in females with desmoid tumors: Updated safety analyses from the DeFi phase 3 study. Cancer, 130(16), 2812–2821. https://doi.org/10.1002/cncr.35324
- Mir, O., Honoré, C., Chamseddine, A. N., Dômont, J., Dumont, S. N., Cavalcanti, A., Faron, M., Rimareix, F., Haddag-Miliani, L., Le Péchoux, C., Levy, A., Court, C., Briand, S., Fadel, E., Mercier, O., Bayle, A., Brunet, A., Ngo, C., Rouleau, E., … Le Cesne, A. (2020). Long-term Outcomes of Oral Vinorelbine in Advanced, Progressive Desmoid Fibromatosis and Influence of CTNNB1 Mutational Status. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 26(23), 6277–6283. https://doi.org/10.1158/1078-0432.CCR-20-1847
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Https://www.nccn.org/professionals/physician_gls (Accessed on October 14, 2020). (n.d.).
- Nishida, Y., Hamada, S., Urakawa, H., Ikuta, K., Sakai, T., Koike, H., Ito, K., Emoto, R., Ando, Y., & Matsui, S. (2020). Desmoid with biweekly methotrexate and vinblastine shows similar effects to weekly administration: A phase II clinical trial. Cancer Science, 111(11), 4187–4194. https://doi.org/10.1111/cas.14626
- Nishida, Y., Tsukushi, S., Shido, Y., Wasa, J., Ishiguro, N., & Yamada, Y. (2010). Successful treatment with meloxicam, a cyclooxygenase-2 inhibitor, of patients with extra-abdominal desmoid tumors: A pilot study. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 28(6), e107-109. https://doi.org/10.1200/JCO.2009.25.5950
- Orbach, D., Brennan, B., Bisogno, G., Van Noesel, M., Minard-Colin, V., Daragjati, J., Casanova, M., Corradini, N., Zanetti, I., De Salvo, G. L., Defachelles, A. S., Kelsey, A., Arush, M. B., Francotte, N., & Ferrari, A. (2017). The EpSSG NRSTS 2005 treatment protocol for desmoid-type fibromatosis in children: An international prospective case series. The Lancet. Child & Adolescent Health, 1(4), 284–292. https://doi.org/10.1016/S2352-4642(17)30045-7
- Penel, N., Chibon, F., & Salas, S. (2017). Adult desmoid tumors: Biology, management and ongoing trials. Current Opinion in Oncology, 29(4), 268–274. https://doi.org/10.1097/CCO.0000000000000374
- Penel, N., Coindre, J.-M., Bonvalot, S., Italiano, A., Neuville, A., Le Cesne, A., Terrier, P., Ray-Coquard, I., Ranchere-Vince, D., Robin, Y.-M., Isambert, N., Ferron, G., Duffaud, F., Bertucci, F., Rios, M., Stoeckle, E., Le Pechoux, C., Guillemet, C., Courreges, J.-B., & Blay, J.-Y. (2016). Management of desmoid tumours: A nationwide survey of labelled reference centre networks in France. European Journal of Cancer (Oxford, England: 1990), 58, 90–96. https://doi.org/10.1016/j.ejca.2016.02.008
- Reitamo, J. J., Scheinin, T. M., & Häyry, P. (1986). The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. American Journal of Surgery, 151(2), 230–237. https://doi.org/10.1016/0002-9610(86)90076-0
- Rigaux, P., Lefebvre-Kuntz, D., Penel, N., & “SOS Desmoïde”. (2015). Pain burden in desmoid tumor patients: A survey of the French Advocacy Group SOS Desmoid. Bulletin Du Cancer, 102(3), 213–216. https://doi.org/10.1016/j.bulcan.2015.02.001
- Saito, Y., Hinoi, T., Ueno, H., Kobayashi, H., Konishi, T., Ishida, F., Yamaguchi, T., Inoue, Y., Kanemitsu, Y., Tomita, N., Matsubara, N., Komori, K., Kotake, K., Nagasaka, T., Hasegawa, H., Koyama, M., Ohdan, H., Watanabe, T., Sugihara, K., & Ishida, H. (2016). Risk Factors for the Development of Desmoid Tumor After Colectomy in Patients with Familial Adenomatous Polyposis: Multicenter Retrospective Cohort Study in Japan. Annals of Surgical Oncology, 23(Suppl 4), 559–565. https://doi.org/10.1245/s10434-016-5380-3
- Shinagare, A. B., Ramaiya, N. H., Jagannathan, J. P., Krajewski, K. M., Giardino, A. A., Butrynski, J. E., & Raut, C. P. (2011). A to Z of desmoid tumors. AJR. American Journal of Roentgenology, 197(6), W1008-1014. https://doi.org/10.2214/AJR.11.6657
- Sinha, A., Hansmann, A., Bhandari, S., Gupta, A., Burling, D., Rana, S., Phillips, R. K., Clark, S. K., & Goh, V. (2012). Imaging assessment of desmoid tumours in familial adenomatous polyposis: Is state-of-the-art 1.5 T MRI better than 64-MDCT? The British Journal of Radiology, 85(1015), e254-261. https://doi.org/10.1259/bjr/42420290
- Skapek, S. X., Ferguson, W. S., Granowetter, L., Devidas, M., Perez-Atayde, A. R., Dehner, L. P., Hoffer, F. A., Speights, R., Gebhardt, M. C., Dahl, G. V., Grier, H. E., & Pediatric Oncology Group. (2007). Vinblastine and methotrexate for desmoid fibromatosis in children: Results of a Pediatric Oncology Group Phase II Trial. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 25(5), 501–506. https://doi.org/10.1200/JCO.2006.08.2966
- Sparber-Sauer, M., Orbach, D., Navid, F., Hettmer, S., Skapek, S., Corradini, N., Casanova, M., Weiss, A., Schwab, M., & Ferrari, A. (2021). Rationale for the use of tyrosine kinase inhibitors in the treatment of paediatric desmoid-type fibromatosis. British Journal of Cancer. https://doi.org/10.1038/s41416-021-01320-1
- Timbergen, M. J. M., van de Poll-Franse, L. V., Grünhagen, D. J., van der Graaf, W. T., Sleijfer, S., Verhoef, C., & Husson, O. (2018). Identification and assessment of health-related quality of life issues in patients with sporadic desmoid-type fibromatosis: A literature review and focus group study. Quality of Life Research: An International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation, 27(12), 3097–3111. https://doi.org/10.1007/s11136-018-1931-3
- Toulmonde, M., Pulido, M., Ray-Coquard, I., Andre, T., Isambert, N., Chevreau, C., Penel, N., Bompas, E., Saada, E., Bertucci, F., Lebbe, C., Le Cesne, A., Soulie, P., Piperno-Neumann, S., Sweet, S., Cecchi, F., Hembrough, T., Bellera, C., Kind, M., … Italiano, A. (2019). Pazopanib or methotrexate-vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): A non-comparative, randomised, open-label, multicentre, phase 2 study. The Lancet. Oncology, 20(9), 1263–1272. https://doi.org/10.1016/S1470-2045(19)30276-1
- Trautmann, M., Rehkämper, J., Gevensleben, H., Becker, J., Wardelmann, E., Hartmann, W., Grünewald, I., & Huss, S. (2020). Novel pathogenic alterations in pediatric and adult desmoid-type fibromatosis – A systematic analysis of 204 cases. Scientific Reports, 10. https://doi.org/10.1038/s41598-020-60237-6
- van Houdt, W. J., Wei, I. H., Kuk, D., Qin, L.-X., Jadeja, B., Villano, A., Hameed, M., Singer, S., & Crago, A. M. (2019). Yield of colonoscopy in identification of newly diagnosed desmoid-type fibromatosis with underlying familial adenomatous polyposis. Annals of Surgical Oncology, 26(3), 765–771. https://doi.org/10.1245/s10434-018-07138-1
- Vitellaro, M., Sala, P., Signoroni, S., Radice, P., Fortuzzi, S., Civelli, E. M., Ballardini, G., Kleiman, D. A., Morrissey, K. P., & Bertario, L. (2014). Risk of desmoid tumours after open and laparoscopic colectomy in patients with familial adenomatous polyposis. The British Journal of Surgery, 101(5), 558–565. https://doi.org/10.1002/bjs.9411
- Walter, T., Zhenzhen Wang, C., Guillaud, O., Cotte, E., Pasquer, A., Vinet, O., Poncet, G., Ponchon, T., & Saurin, J.-C. (2017). Management of desmoid tumours: A large national database of familial adenomatous patients shows a link to colectomy modalities and low efficacy of medical treatments. United European Gastroenterology Journal, 5(5), 735–741. https://doi.org/10.1177/2050640616678150
- Wang, W.-L., Nero, C., Pappo, A., Lev, D., Lazar, A. J., & López-Terrada, D. (2012). CTNNB1 genotyping and APC screening in pediatric desmoid tumors: A proposed algorithm. Pediatric and Developmental Pathology: The Official Journal of the Society for Pediatric Pathology and the Paediatric Pathology Society, 15(5), 361–367. https://doi.org/10.2350/11-07-1064-OA.1